

산업동향
중국 의약품 해외제조원 현장실사에 대한 한국 기업의 대응
- 등록일2018-01-18
- 조회수7365
- 분류산업동향 > 제품 > 바이오의약
-
자료발간일
2018-01-09
-
출처
제약산업정보포털
- 원문링크
-
키워드
#중국#의약품
출처 : 제약산업정보포털
중국 의약품 해외제조원 현장실사에 대한 한국 기업의 대응
KHIDI 상임컨설턴트 / 쑨쉐메이(Sun Xuemei)
▶컨설팅 분야
중국 인허가, 임상
중국 사업개발, 기술 마케팅 등
▶주요 약력
2011-현재 Beijing Lewei Biological & Technology Co. Ltd, Senior Consultant
2009-2011 Shanghai Johnson & Pharmaceuticals, Ltd., Regulatory Affairs Director
2005-2009 Wyeth Pharmaceutical Co. Ltd., Associate Director of Regulatory Affairs
2001-2005 Xian-Janssen Pharmaceutical Ltd., Manager of Regulatory Affairs
1994-1996 Su Zhou Lederle Pharmaceutical Co., Sales Supervisor
1991-1993 HaErbin BinKai Pharmaceutical Co., Director of Pharmaceutical Science Centre
거대 의약품 시장인 중국은 성장 잠재력이 매우 커서 세계 제약회사의 큰 기대를 받고 있다. 오늘날, 제네릭 대국인 중국에서 해외 첨단기술로 개발된 신약, 특히 미충족 수요(Unmet needs) 질병의 치료법이 상당히 인기이다. 개혁 개방 이후 20~30년간 중국 정부는 다양한 혜택으로 해외 제약회사가 중국에서 인허가를 받고 판매할 수 있도록 지원해 왔다. 현재까지 총 4,000여 건의 수입의약품이 허가를 받았고, 이보다 많은 수입의약품의 인허가 신청 건수가 꾸준히 접수되고 있다. 이와 함께, 다양한 규모의 해외 제약회사도 새로운 생산 공정과 밸리데이션 방법 개발에 뛰어들고 있는데, 이는 중국의 관리·감독 기관의 새로운 도전이다.
수입의약품의 해외 제조원의 현장실사는 국가식품약품관리감독총국(China Food and Drug Administration, CFDA)의 관리·감독 항목에서도 큰 도전이다. 대규모의 재원이 필요하고 실사 인력의 기술과 지식의 폭에 대해서도 훨씬 높은 수준을 요구하기 때문이다. 실사 전문가는 해외의 품질관리 개념을 이해하고 전문성을 갖추어야 하는 동시에 우수한 외국어 소통 능력과 문화교류 능력을 갖추어야 한다. 사실 이 같은 조건을 갖춘 인재는 매우 드물다. 현재까지 중국 CFDA가 수입의약품의 해외 제조원 현장 실사를 매우 낮은 빈도로 실시하여 일부 제조사가 요행수를 바라는 경우도 있었다. 또한, 규정에 따른 관리를 소홀히 하기도 하고, 제조 및 밸리데이션에 중대 변경 사항이 발생 했을 때 의무사항인 보완 신청도 적극적으로 하지 않았다. 그 결과 CFDA가 수입의약품의 상황을 적시에 파악하지 못하여 중국 환자의 의약품 사용에 있어서 위험성이 증가했다. 2013년에서 2017년까지 CFDA가 직속 산하기관인 식품약품심사검증센터(Center for Food and Drug Inspection, CFDI)에 권한을 부여하여 제한된 여건에서지만 해외 제조원에 대한 실사를 시작했다. 먼저 바이오제제, 무균제제(주사제, 안약, 흡입제, 삽입제), 및 중약(中?)과 천연물에 제품 등 고위험 제품부터 엄격하게 생산실사를 진행했는데 그 가운데 다음의 제품이 수입 금지를 명령을 받았다.


입의약품 제조원에 대한 현장 실사 관리 규정
- 의약품의 해외 제조원에 대한 실사 작업은 CFDA가 진행하는 인허가 평가, 심사 중 또는 이미 품목허가를 받은 수입의약품의 생산현장에 대한 실사이다. 수입의약품의 관리·감독 강화, 의약품 수입 자체의 규범 마련, 수입의약품의 품질과 안전 보장에 그 목적이 있다.
- 수입의약품의 제조원 실사는 CFDA가 발급한 〈수입약품등록증(?口?品注??)〉 또는 〈의약품등록증(???品注?? )〉을 득한 모든 수입의약품 생산 기업 및 현재 CFDA에 〈수입약품등록증(?口?品注??)〉 또는 〈의약품등록증(???品注?? )〉를 신청 중인 해외 의약품 제조기업에 적용된다.
- CFDA은 인허가 심사, 일상적 관리·감독, 항구 검역 및 민원 등 각종 정보에 따라 현장 실사를 받는 수입의약품 기업과 제품 명단을 확보하고 시간 등 이와 관련된 정보를 수입의약품 생산기업의 중국 국내 사무실이나 위탁 대행 기업(이하 대행 기관)에 사전 통보한다.
- 대행기관은 수입의약품 생산기업과의 연락과 CFDA의 의약품인증관리센터에 즉각적인 관련 필요자료 제출을 책임진다.
- 실사 예정인 수입의약품 제조원은 실사 기간에 실사를 받을 제품을 로트 단위로 생산해야 한다.
-현장 실사는 팀장 책임제로 진행한다. 실사팀은 일반적으로 2~5명의 실사관으로 구성한다.
-현장실사의 첫 회의는 실사팀의 팀장이 주재하고 실사 범위, 실사 일정 및 기업 수행 인원을 결정한다. 실사 기율(?律) 및 주의 사항을 발표한다.
-현장실사의 주요 내용은 다음이다. 의약품 인허가 신고자료, 현장 자료와 생산과정의 일치성, 의약품 생산 과정과 《의약품 생산 품질 관리 규범(2010년 수정)(?品生??量管理?范(2010年修?)》 부합 여부 등
-실사 받는 수입의약품 기업을 실사에 필요한 관련 자료를 즉시 제공해야 한다. 실사 작업의 필요에 따라 실사관이 사진 및 동영상 촬영 등의 방법으로 증거를 수집할 수 있다. 기업이 사진 또는 동영상 촬영을 거부할 경우 실사관은 관련 상황을 보고서에 상세히 기재해야 한다. 필요할 경우 실사관은 샘플 일부를 가져와 중국 국내에서 시험을 진행할 수 있다.
-현상 실사가 종료되면 실사팀은 현상실사 상황을 분석 및 종합하여 기업의 부족한 점을 확인한다. 분석 및 자료를 종합하는 기간에 기업의 수행인은 참석하지 않는다.
-마지막 회의에서 실사 팀은 실사를 받는 수입의약품 제조원에 실사 중에 발견한 지적사항을 구두로 알린다. 이에 대해 실사를 받은 수입의약품 제조원이 이의를 제기할 경우 그에 대해 설명하고, 필요 할 경우 관련 상황을 추가 확인하고 결과에 따라 해당 지적사항을 수정한다.
-실사 종료일로부터 2개월 안에 CFDA의 의약품인증관리센터가 서면으로 현장실사 보고서를 대행기관에 전달한다. 실사에서 명확한 지적사항이 없거나 지적사항을 즉시 바로 잡을 수 있는 기업에 대해서는 CFDA의 의약품 인증관리센터가 서면으로 현장실사 보고서를 대행기관에 다시 보내지 않고 바로 15조 관련 규정에 따라 처리한다. 대행기관은 현상실사 보고서를 받은 후 1개월 안에 실사받은 기업의 보고서를 시정하여 CFDA 의약품 인증관리센터에 제출한다. 특수 상황으로 기한 내에 시정 보고서를 제출 할 수 없을 경우 CFDA 의약품 인증센터에 제출 날짜 변경 신청하고 시간을 확정한다. 단, 1개월 이상 연장할 수 없다.
-CFDA의 의약품인증관리센터 약품인증관리센터는 대행기관이 제출한 시정 상황 보고서를 접수한 후 1개월 안에 위험관리 원칙에 따라 실사 상황에 대해 종합적으로 평가한다. 실사 결과는 ‘적합’,’ 시정 후 적합’, ‘부적합’으로 나뉜다. 판단 원칙은 다음과 같다.
1. 의약품 제조 및 품질 제어와 신청자료가 일치하고 의약품 GMP 요구조건에 따라 제조되었으면 ‘적합’으로 판정한다.
2. 현장실사에서 여러 주요 지적사항이 발견되었을 경우, 제출한 시정보고서에 지적을 시정 후 의약품 GMP 요구 사항에 따라 제조할 수 있으면, ‘시정 후 적합’으로 판정한다.
3. 현장실사에서 과장 행위 또는 품질에 영향을 미칠만한 핵심 사안과 신고 자료가 일치하지 않은 경우, 중대 지적사항 또는 여러 주요 지적사항에 대해서 실사받은 제조원이 의약품 GMP 요구조건에 따라 생산할 수 없을 경우 실사 결과를 ‘부적합’으로 판정한다.
-‘적합’, 시정 후 적합’으로 결론 난 기업에 대해 CFDA는 실사 결론 발표 후 1개월 안에 대행기관에 서면으로 피드백을 한다.
-‘시정 후 적합’으로 나온 기업에 대해 CFDA는 대행기관에 《경고장(警告信)》을 발송하고 해당 의약품의 수입 잠정 중단을 명령하거나 ,차후 현장 실사에서 적합 판정을 받을 때까지 해당 의약품의 인허가 평가와 심사를 잠정 중단한다. 이와 함께 각 항구의 CFDA에 통보하여 해당 의약품의 《수입의약품 통관(?口?品通??, Drug Import Customs Clearance)》을 잠시 중단한다.
-이미 수입된 의약품의 경우 CFDA는 상황의 경중에 따라 기업에 의약품의 리콜을 명령하거나 기타 처리 방법을 결정한다.
해외 제조원에 대한 현장실사 항목은?
1. Content of Site Master File
??主文件??
1.1 Content of Site Master File
??主文件??
- Name and official address of the manufacturer;
企?名?、注?地址;
- Names and street addresses of the site, buildings and production units located on the site;
企?生?工?以及工??建筑及生???名?和地址;
- Contact information of the manufacturer including 24 hrs telephone number of the contact personnel in the case of product defects or recalls;
企??系方式(包括出??品缺陷或召回事件?24小??系人??);
- Identification number of the site as e.g. GPS details, D-U-N-S (Data Universal Numbering System) Number (a unique identification number provided by Dun& Bradstreet) of the site or any other geographic location system.
??????,例如GPS??情?,D-U-N-S??(?据通用??系?)(一?由Dun& Bradstreet提供的?特????)或者任何其他地理定位系?。
1.2 Authorised pharmaceutical manufacturing activities of the site.
?品生??可范?
- Copy of the valid manufacturing authorization issued by the relevant Competent Authority in Appendix 1; or when applicable, reference to the EudraGMP database. If the Competent Authority does not issue manufacturing authorizations, this should be stated;
附件1中提供相??管机???的有效生??可文本?印件,必要?,可?考EudraGMP?据?。如遇?管机?不??生??可情?,?予以?明。
- Brief deion of manufacture, import, export, distribution and other activities as authorised by the relevant Competent Authorities including foreign authorities with authorized dosage forms/activities, respectively; where not covered by the manufacturing authorization;
?要描述由相??管机??可的生?、?口、出口、分?和其他活?,包括?可文件中?有提及的?外机??可的?型/生?活?等;
- Type of products currently manufactured on-site (list in Appendix 2) where not covered by Appendix 1 or the EudraGMP database;
在附件2中列出附?1或EudraGMP?据?中?有提及的工?目前生?的?品?型;
- List of GMP inspections of the site within the last 5 years; including dates and name/country of the Competent Authorities having performed the inspection. A of current GMP certificate (Appendix 3) or reference to the EudraGMP database should be included, if available.
近5年工?接受GMP??情?,包括????和?施??的?管机?名?及?家。如果有,?在附件3中提供?前的GMP??的?印件或?考EudraGMP?据?。
1.3 Any other manufacturing activities carried out on the site
工?目前?行的其?生?活?
- Deion of non-pharmaceutical activities on-site, if any. 如工?有非?品生?活?,??明。
2. Quality Management System of the Manufacturer
生?企??量管理?系
2.1 The quality management system of the manufacturer
生?企??量管理?系
- Brief deion of the quality management systems run by the company and reference to the standards used;
?要描述公司?量管理?系?行情?以及?考的?准;
- Responsibilities related to the maintaining of quality system including senior management;
包括高?管理?在?的?量?系相???,。
- Information of activities for which the site is accredited and certified, including dates and contents of accreditations, names of accrediting bodies.
工??量?系?得???可的情?,包括???可日期、?可?容、?可机?名?等。
2.2 Release procedure of ed products
成品放行程序
- Detailed deion of qualification requirements (education and work experience) of the Authorised Person(s)/ Qualified Person(s) responsible for batch certification and releasing procedures;
??描述??批???放行程序的授?人的??要求;
- General deion of batch certification and releasing procedure;
?述批???放行程序;
- Role of Authorised Person/ Qualified Person in quarantine and release of ed products and in assessment of compliance with the Marketing Authorisation;
授?人/?品放行人在待??放行以及上市?可一致性??中的??;
- The arrangements between Authorised Persons/ Qualified Persons when several Authorised Persons/ Qualified Persons are involved;
?涉及多名授?人?的工作安排;
- Statement on whether the control strategy employes Process Analytical Technology (PAT) and/or Real Time Release or Parametric Release.
??明是否?用?程分析技?(PAT)及??或??放行?品。
2.3 Management of suppliers and contractors
供?商和合同商的管理
- A brief summary of the establishment/knowledge of supply chain and the external audit program;
?述公司供??以及外部???目等情?;
- Brief deion of the qualification system of contractors, manufacturers of active pharmaceutical ingredients (API) and other critical materials suppliers;
?述合同商、原料?生?企?及其他??物料供?商的????系?;
- Measures taken to ensure that products manufactured are compliant with TSE(Transmitting animal spongiform encephalopathy)guidelines.
采取?些措施?保生?品?符合TSE (?物?染?海??病)指南要求。
- Measures adopted where counterfeit/falsified products, bulk products(i.e.unpacked tablets), active pharmaceutical ingredients or excipients are suspected or identified.
?假?以及原?料等造假???高的地?,采取?些措施予以控制。
- Use of outside scientific, analytical or other technical assistance in relation to manufacture and analysis;
委托生?和委托??及其??目委托情?
- List of contract manufacturers and laboratories including the addresses and contact information and flow charts of supply-chains for outsourced manufacturing and Quality Control activities; e.g. sterilization of primary packaging material for aseptic processes, testing of starting raw materials etc, should be presented in Appendix 4;
合同生?企?和??室名?,包括以下信息:地址、?系方式、委托生?和?量??活?的供??流程?;例如:无菌工??品所用?包?材料?菌、起始物料的??等均?在附?4中予以表述?楚;
- Brief overview of the responsibility sharing between the contract giver and acceptor with respect to compliance with the Marketing Authorisation (where not included under 2.2).
?述委托方和受托方在?品放行中的?任(不包括在2.2中)。
2.4 Quality Risk Management (QRM)
?量??管理(QRM)
- Brief deion of QRM methodologies used by the manufacturer;
?述企??量??管理方法
- Scope and focus of QRM including brief deion of any activities which are performed at corporate level, and those which are performed locally. Any application of the QRM system to assess continuity of supply should be mentioned.
按公司不同??(集?和生??)?述?量??管理的范?和重点,包?提及任何??供?持?性的?量??管理?系?用。
3. Personnel 人?
- Organisation chart showing the arrangements for quality management, production and quality control positions/titles in Appendix 5, including senior management and Authorised Person(s) / Qualified Person(s);
企??量管理、生?和?量控制及其??人的??机??,包括高?管理?和授?人等(附件5);
- Number of employees engaged in the quality management, production, quality control, storage and distribution respectively.
?事?量管理、生?、?量控制、?存及分?的?工?量。
4. Premises and Equipment ?房和??
4.1 Premises
?房
- Short deion of plant; size of the site and list of buildings. If the production for different markets, i.e. for local, EU, USA, etc. takes place in different buildings on the site, the buildings should be listed with destined markets identified (if not identified under 1.1);
?述生?工?情?,包括?地面?和各建筑物名?等,如不同建筑物生?的品?面向?地以及?盟、美?等不同市?,?在特定市?的建筑物上注明(如未在1.1明?)
- Simple plan or deion of manufacturing areas with indication of scale (architectural or engineering drawings are not required); Lay outs and flow charts of the production areas (in Appendix 6) showing the room classification and pressure differentials between adjoining areas and indicating the production activities (i.e.compounding, filling, storage, packaging, etc.) in the rooms;
?述生??域?模情?,附???平面布局?、生??域的平面布局?和流向?,?明比例(不需要建筑或工程??)。???注出房?的????、相?房?的?差,?且能指示房?所?行的生?活?(例如:配料、灌?、?存、包?等)(附件6);
- Lay-outs of warehouses and storage areas, with special areas for the storage and handling of highly toxic, hazardous and sensitising materials indicated, if applicable;
??和?存?域的平面?,如果有,包括?存和?理高毒性、危?性?敏感物料的特殊?域。
- Brief deion of specific storage conditions if applicable, but not indicated on the lay-outs.
如有,??述特殊?存?件情?,但不需在平面?上注明。
4.1.1 Brief deion of heating, ventilation and air conditioning (HVAC) systems
?述空??化(HVAC)系?
- Principles for defining the air supply, temperature, humidity, pressure differentials and air change rates, policy of air recirculation (%).
?述空??化系???原?,如送?、?度、?度、?力差以及??次?、回?等 (%)。
4.1.2 Brief deion of water systems
?要描述水系?
- Quality references of water produced;
水????准
- Schematic drawings of the systems in Appendix 7.
水系?示意?附?7
4.1.3. Brief deion of other relevant utilities, such as steam, compressed air, nitrogen, etc.
?要描述其?相?公用?施,例如蒸汽、??空?、??等系?。
4.2 Equipment
??
4.2.1 Listing of major production and control laboratory equipment with critical pieces of equipment identified should be provided in Appendix 8.
列出生?和??用主要?器、??附?8。
4.2.2 Cleaning and sanitation
???消毒
- Brief deion of cleaning and sanitation methods of product contact surfaces (i.e. manual cleaning, automatic Clean-in-Place, etc).
?述??品直接接???、工器具的表面?洗、消毒方法及??情?(例如:人工??、自?在???等)。
4.2.3 GMP critical computerised systems
??品生??量相?的???算机化系?
- Deion of GMP critical computerised systems (excluding equipment specific Programmable Logic Controllers (PLCs)).
?述??品生??量相?的??的?算机化系?情?(不包括???程器(PLCs))。
5. Documentation
文件
- Deion of documentation system (i.e. electronic, manual);
描述企?的文件系?(例如?子、??);
- When documents and records are stored or archived off-site (including pharmacovigilance data, when applicable): List of types of documents/records; Name and address of storage site and an estimate of time required retrieving documents from the off-site archive.
如文件和??在生?工?外保存(如有,包括?物警戒?据),?提供外存的文件/??目?、?存?所的名?和地址以及???外取回文件所需的??。
6. Production
生?
6.1 1.1 Type of products
?品?型
(references to Appendix 1 or 2 can be made):
(可?考附件1或2):
- Type of products manufactured including
生?品??型
? list of dosage forms of both human and veterinary products which are manufactured on the site
工?生??型一?表(包括人用??用?品)
? list of dosage forms of investigational medicinal products (IMP) manufactured for any clinical trials on the site, and when different from the commercial manufacturing, information of production areas and personnel
工?生??床??用?品(IMP)?型一?表,如生??所?上市生?品?不同,?提供生??域和生?人?信息。
- Toxic or hazardous substances handled (e.g. with high pharmacological activity and/or with sensitising properties);
毒性或危?物?的?理情?(如高活性和/或高致敏?品);
- Product types manufactured in a dedicated facility or on a campaign basis, if applicable;
如有,??明?用??或?段生?制造?品情?;
- Process Analytical Technology (PAT) applications, if applicable: general statement of the relevant technology, and associated computerised systems.
如有,??明?程分析技?(PAT)?用情?,??述相?技?和?算机化系??用情?。
6.2 Process validation
工???
- Brief deion of general policy for process validation;
?要描述工???的原?;
- Policy for reprocessing or reworking.
返工或重新加工的原?。
6.3 Material management and warehousing
物料管理和??
- Arrangements for the handling of starting materials, packaging materials, bulk and ed products including sampling, quarantine, release and storage;
起始物料、包?材料、半成品?成品的?理,包括取?、待?、放行??存;
- Arrangements for the handling of rejected materials and products.
不合格物料和?品的?理
7. Quality Control (QC) ?量控制
- Deion of the Quality Control activities carried out on the site in terms of physical, chemical, and microbiological and biological testing.
描述理化??、微生物及生物???等?量控制活?。
8. Distribution, Complaints, Product Defects and Recalls
分?、投?、?品缺陷?召回
8.1 Distribution (to the part under the responsibility of the manufacturer)
分?(?于制造商???的部分)
- Types (wholesale licence holders, manufacturing licence holders, etc) and locations (EU/EEA, USA, etc.) of the companies to which the products are shipped from the site;
分?商?型(包括是否持有???可?或制造?可?等)及其所在地?(?盟/?洲???、美?等);
- Deion of the system used to verify that each customer / recipient is legally entitled to receive medicinal products from the manufacturer;
描述用????客/接受者的系?,以?明?客有合法?格接收?品;
- Brief deion of the system to ensure appropriate environmental conditions during transit, e.g. temperature monitoring/ control;
?要描述?品在???程中?保其符合?存?件要求的的措施,例如: ?度??/?控;
- Arrangements for product distribution and methods by which product traceability is maintained;
?品分?管理以及?保其可追踪的方法。
- Measures taken to prevent manufacturers’ products to fall in the illegal supply chain
防止?品流入非法供??的措施。
8.2 Complaints, product defects and recalls
投?、?品缺陷?召回
- Brief deion of the system for handling complains, product defects and recalls.
?要描述投??理、?品缺陷?召回系?。
9. Self Inspections
自?
Short deion of the self inspection system with focus on criteria used for selection of the areas to be covered during planned inspections, practical arrangements and follow-up activities.
?要描述企?自?系?,重点?明自???中涉及范?的???准、自??施以及整改情?。
相?附件 :
- Appendix 1 Copy of valid manufacturing authorisation
附件1 有效的制造?可文件?印件
- Appendix 2 List of dosage forms manufactured including the INN-names or common name (as available) of active pharmaceutical ingredients (API) used
附件2 所有生??型目?,包括所用原料?的INN名?或通用名(如有)
- Appendix 3 Copy of valid GMP Certificate
附件3 有效的GMP???印件
- Appendix 4 List of contract manufacturers and laboratories including the addresses and contact information, and flow-charts of the supply chains for these outsourced activities
附件4 合同生?企?和??室情?一?表,包括地址和?系信息以及外包活?的供??流程?。
- Appendix 5 Organisational charts
附件5 ??机??
- Appendix 6 Layouts of production areas including material and personnel flows, general flow charts of manufacturing processes of each product type (dosage form)
附件6 生??域平面?,包括物料和人?流向?,各?型(?型)?品 生?工?流程?
- Appendix 7 Schematic drawings of water systems
附件7 水系?示意?
- Appendix 8 List of major production and laboratory equipment
附件8 ??生??????室??、?器??
해외 현장실사에서 발견된 주요 문제점
2016년 해외 실사에서 나타난 지적사항 분석
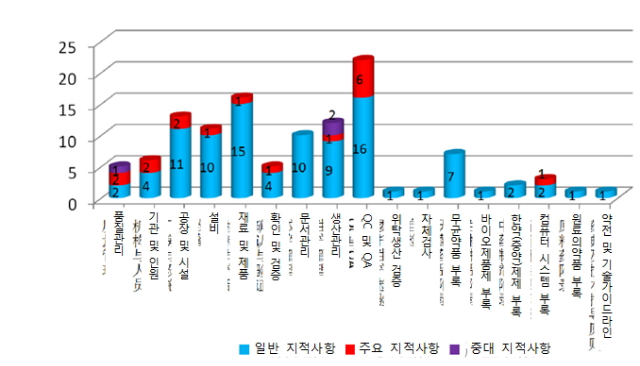
현장실사의 주요 지적사항 및 사례
1. 실제 생산 공정, 생산 현장 등이 신고자료와 일치하지 않고 중대한 변경 사항 등을 중국에 신고하지 않은 채로 현장에 적용되었을 경우
사례: 한 수입 제품의 실제 생산 공정이 중국에 재등록(2014년)된 생산 공정과 일치하지 않았음. 2001년 유럽의 한 의약품 당국 현장실사에서 해당 회사의 생산 공정이 신고내용과 일치하지 않아 실제 상황에 맞추어 생산공정을 신고하도록 요구하였음. 2002년 주관 의약품 당국은 해당 공정을 정식으로 허가하였음. 그러나 해당 기업은 CFDA에 보충신청을 하지 않았고, 2014년 재등록 時 변경 사실을 고의로 은폐하여 최초 허가 받았던 내용으로 신고했음. 2015년 CFDA가 해당 제품에 대해 현장실사를 진행했을 때 위의 문제를 발견하고 중국으로의 수입과 중국 국내 판매를 즉시 정지했음. 이미 중국에서 유통된 제품도 리콜하였음.
2. 데이터 신뢰성에 중대한 문제가 있고 제품 품질에 심각한 영향 미침.
데이터 신뢰성 문제는 분석 실험실에만 국한되는 것이 아님. 실사관은 생산구역 및 미생물 실험실까지 관련 기록을 찾아봄. 사례 1: 생산 구역에서 제품을 생산할 때 신고하지 않은 로트 생산 단위 기록은 데이터 신뢰성의 문제임. 이는 로트 기록은 일괄운영처리를 시작할 때 기록하기 때문임. 실사관이 실사 과정에서 이미 한 로트가 생산되었으나 이 기록이 없고, 작업자가 로트 생산 후 사무실에서 모든 관련 내용을 다시 작성했다는 것을 발견했음.
사례2: 화학실험실에서 고속능액체크로마토그래피(모델명: SHIMADEU PROMINANCE, 기기번호: HPLC No.5)를 검사한 결과 실사관끼리 공유하는 비밀번호로 데이터처리시스템에 접속하여 보니, 해당 기기의 감사추적기능 방법이 바뀐 것만 기록되었고 전체 실행 테이터는 기록되지 않았음을 발견함.
3. 회사의 출시 조건(Release criteria)이 중국에서의 등록 인허가 조건과 일치하지 않은 경우.
사례 1: 한 수입 바이오제제가 2000년에 수입 인허가 신청할 때 수입기준 확인기관이 해당제품의 수입기준을 수정하였음. 미생물한도시험을 무균시험으로 변경한 것이 주요 내용임. 2016년 현장실사에서 해당 회사가 여전히 미생물한도시험을 시행하는 것이 발견되었음.
사례2: 기업의 정제수 시험을 유럽 기준에 따라 시행하고 중국 약전 2015년 버전(2015版)》에 따라 시행하지 않음.
이상의 문제는 모두 CFDA 해외 현장실사에서 발견된 공통적인 문제였다. 한국 기업도 이 같은 문제에 충분히 주목하기를 바라는 마음으로 위의 내용을 공유한다. 이미 중국에 출시된 제품은 반드시 제때에 변경 신청을 진행하고, 현재 수입의약품 등록 중인 제품은 중국 관련 법 변화를 관찰하여 해당 법규와 기준을 준수하는 것이 바람직하다.
...................(계속)
☞ 자세한 내용은 내용바로가기 또는 첨부파일을 이용하시기 바랍니다.
지식
동향