

기술동향
Microarrays for Gene-Expression Studies
- 등록일2009-08-14
- 조회수9096
- 분류기술동향
-
자료발간일
2009-08-01
-
출처
GEN
- 원문링크
-
키워드
#Gene#Gene-Expression#Microarrays
Microarrays for Gene-Expression Studies
Cost-Effective Alternative to Sequencing Boasts a Multitude of Applications
Elizabeth Lipp
While microarray technology is broadening its applications in genomics and proteomics, the technique remains a vibrant contributor in the area of gene expression. Much of the overall developmental effort is directed at making microarrays more high throughput in both genome coverage and in sample numbers.
Victor Levenson, Ph.D., associate professor at Rush University Medical Centre, will review available platforms for microarray-based methylation detection and discuss their advantages and limitations for biomarker development at Select Biosciences’ “Microarray World Congress” to be held this month.
Dr. Levenson will also discuss correlative biomarkers for early diagnosis, prediction of outcome, and response to drugs. “There are different technologies for tumor detection based on tumor-specific mutations, but it is rather difficult to find a rare mutation, especially if we do not know what genes are mutated,” he says. “Instead, we examine tumor-specific abnormalities in DNA methylation.”
Reaching the tumor tissue to extract its DNA can be difficult. “But tumor DNA actually floats in blood, which is a treasure trove for biomarker development,” Dr. Levenson adds. “How tumor DNA makes its way into blood is only partially understood, but we can use it to develop markers for tumor detection. Importantly, the whole genome of the tumor is in blood, so multiple genes can be analyzed in the same sample for an extremely accurate result. That is why we look for tumor-specific patterns of methylation rather than a change in methylation of a single gene.”
The need to determine specific patterns requires analysis of multiple genes in DNA from the same sample, and that brings us to microarrays and how they work, notes Dr. Levenson. “We use microarrays to screen for differences in a multitude of genes, and select the most informative. Once that is done, only informative genes will be necessary to detect the disease, and microarrays will no longer be needed for a clinical test. Once we select the most informative genes-those that give us the best diagnosis-we will test 5?10 genes per disease by a simple PCR.”
To select these informative genes, however, microarrays are essential. “The Illumina GoldenGate platform can test more than 800 genes, but depends on bisulfite modification, so each analysis requires a lot of DNA,” Dr. Levenson continues.
“An alternative platform developed by Roche NimbleGen can analyze up to 28,000 promoters in each sample, but detection of methylation is based on immunoprecipitation, so the starting amount of DNA again creates the bottleneck. In our lab, we use a custom-made microarray with 56 genes for the proof-of-principle studies. Once this phase is over, biomarker production runs will employ a 12,000 gene microarray. Upstream processing involves digestion of isolated DNA with a methylation-sensitive restriction enzyme, so the whole test requires only 0.3?0.5 ng of DNA.”
There are two major ways to prepare a microarray-you can spot synthetic oligos on a special support (usually glass slides) or you can synthesize the oligo directly on the support. “These spotted microarrays allow directional attachment of oligos,” notes Dr. Levenson. “For example, arrays produced for us by Microarrays have oligos with linkers at the 5-prime end, so the oligo is fixed to the glass support in one predetermined orientation.” Not all spotted arrays are like that-if unmodified DNA is simply placed on support it will attach randomly, which might reduce its ability to hybridize.
Spotted arrays can be made by contact or noncontact spotting. The first depends on a physical touch (e.g., when needle-based instrument deposits oligos using very thin needles); the noncontact method applies electrical discharge or inkjet-type technology. Density of spots is limited in both cases-by physical thickness of a needle for contact printers or by the size of the aperture for inkjets. This means that the spot diameter can’t be smaller than a certain limit, so their number on each slide is also limited.
To increase spot density in Affymetrix arrays oligos are synthesized directly on solid support by photolithography, so that literally millions of oligos are positioned on each mictoarray. This comes at a cost-accuracy of synthesis is good for short oligos but insufficient for longer molecules. “Despite these limitations, only Affymetrix makes tiling microarrays that cover the whole genome with very small gaps between two adjacent probes,” says Dr. Levenson. “For our work, however, these arrays are far too complicated, so spotted arrays are the most appropriate.”
Biomarkers for Colon Cancer
Characterizing DNA methylation patterns is a hot topic, says Sungwhan An, Ph.D., president and CEO at Genomictree, who will also be presenting at the meeting. “We established a method for methyl DNA isolation assay (MeDIA) using the binding property of methyl-DNA binding polypeptide to methyl DNA.
“We also identified promising methylation targets for early detection and prediction of disease recurrence of colon cancer using MeDIA-coupled CpG microarray. Basically, we isolated methylated DNA specifically from a pool of fragmented genomic DNA with methyl DNA-binding domain and performed hybridization after labeling.”
Dr. An says that his company does a lot of private sector work in the microarray space, and the identification of methylation biomarkers is of great interest.
“Eventually, our goal is to identify biomarkers for colon cancer-certain methylation biomarkers for early detection and stratification,” says Dr. An. “We generated a recombinant histidine-tagged minimum size of methyl-DNA binding domain and used nickel magnetic particles for methyled DNA enrichment and adopted freeze-drying for preserving protein. This provides us better conditions for experimental replication and specificity and shelf life.
“We have a nice study design comparing genome-wide methylation patterns in a primary tumor and paired nontumor tissues of patients with colon cancers, half of them developed disease-recurrence and two super-normal tissues of healthy individuals over common reference DNA as a role of internal control.
“Methylation patterns were discerning enough to distinguish a primary tumor from nontumor tissues, which was closely grouped to super-normal tissues by unsupervised hierarchical clustering. We wanted to see if the stratification was prominent enough, and we found that this method of stratification worked on a small scale.”
Dr. An says that using methylation to study DNA is helping scientists make great inroads in cancer research. “RNA gives good feedback, but DNA is more stable for methylation.”
Characterizing Threats
Another application of microarrays that will be explored is the characterization of emerging, unknown, and engineered threats. Crystal Jaing, Ph.D., group leader at the Lawrence Livermore National Laboratory biosciences and biotechnology division will discuss some of the work that her group is doing with microarrays in the area of biodefense. “Our team is strong on bioinformatics, and microarrays play a big part in our research,” she notes.
PCR is limited in the number of pathogens it can analyze. “You can multiplex PCR to detect a number of pathogens, but it does not provide the throughput to analyze all possible pathogens present from one assay,” Dr. Jaing says.
She reports that there are several applications of microarrays at work. The first involves using microarray probes developed to find viruses and bacterial agents. “The key capability is the comprehensive viral and bacterial sequences we have covered in our microarray to enable rapid metagenomic analysis of environmental and clinical samples.
“The second microarray targets microbial antibiotic resistance and virulence mechanisms. By targeting virulence gene families as well as genes unique to specific biothreat agents, these arrays will provide important data about the pathogenic potential and drug-resistance profiles of unknown organisms in environmental samples. This array can provide value to the Biowatch surveillance programs by providing rapid additional characterization of positive samples.”
There are many advantages to using microarrays in her field, but Dr. Jaing acknowledges some challenges as well. “Working with microarrays from the bioinformatics standpoint presents a continual challenge in that there are always new viruses and bacteria to keep up with, thereby, constantly needing to update the database. Though microarray prices are coming down, not every lab has the capability. It’s not like PCR, where you find it in every lab.”
“Sequencing still provides the highest resolution, but it’s also expensive. Microarrays provide a cost-effective way to screen samples quickly, although I have to say that all three technologies work well together.”
Ecological Applications
Researchers at Indiana University are using gene-expression approaches to evaluate the use of Daphnia to study the effects of chemical threats on the environment.
(Paul Hebert, Biodiversity Institute of Ontario)
(Paul Hebert, Biodiversity Institute of Ontario)
“There is always a lot of discussion about high-throughput sequencing versus microarrays for gene expression,” says John Colbourne, Ph.D., genomics director at Indiana University Center for Genomics and Bioinformatics. For instance, Dr. Colbourne reports that the data from Roche NimbleGen tiling-path arrays is comparable to RNA sequencing with the Illumina Genome Analyzer.
Yet, for targeted studies that screen transcriptional changes across large numbers of conditions and for large numbers of samples, practicality becomes a central issue. “A main advantage that we see in using microarrays is that multiplexing 12 to 24 samples on a single chip can save time and money as we move toward understanding transcriptional variation at the level of populations.”
Studies in Dr. Colbourne’s lab encompass the fields of evolutionary ecology, molecular toxicology, systematics, and functional genomics. His lab is now concentrating more on environmental issues and applying approaches traditionally used for gene expression in cancer research to environmental studies.
“Using microarrays, we hope to be running hundreds upon hundreds of experiments in a week,” reports Dr. Colbourne. “The limitation is that there are not enough spaces for independent trials on a single chip. NimbleGen has 12 plexes and is looking to go to 24 plexes and beyond, not just to increase single throughput but to also decrease expenses and to minimize experimental variation. Microarrays can interrogate gene profiles such that multiple samples can be processed discretely.”
Dr. Colbourne and his team seek to connect gene expression and genome structure with individual fitness and population-level responses to environmental chemicals and natural stressors. His work revolves primarily around Daphnia as a model organism to discover the effects of chemical threats to the environment.
“There are currently 80,000 chemicals in the environment, with an additional 2,000 introduced every year,” Dr. Colbourne says. “Less than seven percent of these get tested. We chose Daphnia because it is already a model system for monitoring the health of freshwater ecosystems. It’s a big push to discover how these genomic systems in the environment are affected by the chemicals-often, we don’t know if a chemical is safe or not. There is no systematic and inexpensive way to test the safety of chemicals in environmentally relevant conditions. Chemicals are mostly found in combinations, which is problematic, as they will interact in unexpected ways to affect the health of any animal.”
Dr. Colbourne says that Daphnia is one of the best-characterized genomic systems. “But in 25?30 percent of any genome, you have known genes with unknown functions. Chances are these genes have condition-specific regulation in natural settings, and therefore, no effects are ever detected in the laboratory. When we succeed in routinely studying gene expression within natural populations and ecosystems, we will likely discover a much greater diversity of gene functions, and perhaps even large transcribed areas of genomes that are currently unannotated.”
Dr. Colbourne says that Daphnia is one of the best-characterized genomic systems. “But in 25?30 percent of any genome, you have known genes with unknown functions. Chances are these genes have condition-specific regulation in natural settings, and therefore, no effects are ever detected in the laboratory. When we succeed in routinely studying gene expression within natural populations and ecosystems, we will likely discover a much greater diversity of gene functions, and perhaps even large transcribed areas of genomes that are currently unannotated.”
Dr. Colbourne believes that there are practical reasons to use microarrays in this way. “Regulatory agencies are anxious for new technologies that are more sensitive, less expensive, and better able to assess the potential risk of environmental chemicals to humans and to the environment. With sufficient funding to produce the required reference gene-expression databases, we should soon begin monitoring the health of Daphnia populations using microarrays, which will simultaneously diagnose the presence of chemicals in regional waters and their health effects. There are two uses of microarrays: as a chemical detection tool on sentinel species within ecosystems, and as a method to classify environmental risk based on knowing toxicological effects.”
Reverse Engineering
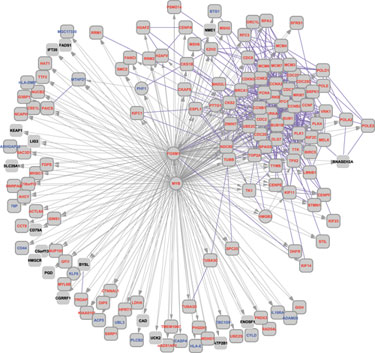
MYB/FOXM1 network from the Human B Cell Interactome (HBCI)
“The work that we do with microarrays is quite different from much of the field,” says Andrea Califano, Ph.D., professor in the department of biomedical informatics at Columbia University Medical Center. “We use them to reverse engineer the regulatory network controlling cell behavior and then to identify master regulators of specific phenotypes. Instead of looking for genes that are differentially expressed, we associate regulatory changes in the cell with genes that control them. This typically requires analyzing hundreds of samples at a time.”
The end result is that Dr. Califano’s lab has assembled genome-wide, context-specific maps of molecular interactions in human cells by integrating several reverse-engineering approaches, which include the use of microarrays. “We have a good repertoire of control mechanisms of the cell, including transcriptional, post-transcriptional, and post-translational ones,” notes Dr. Califano, “We use microarrays to define cellular transition, and to have a complete map of regulatory changes in the cell. These resulting maps have shown significant promise in the rational elucidation of both physiological and pathological phenotypes.”
Their approach, using microarray data to reverse engineer the regulatory process of the cell, makes it possible to infer robust prognostic markers by identifying upstream master regulators that are causally related to the presentation of the phenotype of interest. “You couldn’t do this with sequencing at this stage because profiling hundreds of samples by deep sequencing is still prohibitively expensive,” Dr. Califano adds. “We have, however, successfully used deep sequencing for short regulatory RNA profiling.”
-
다음글
- 다음글이 없습니다.
-
이전글
- 이전글이 없습니다.
관련정보
지식