

기술동향
Proving the protein-only hypothesis in neurodegeneration
- 등록일2012-07-10
- 조회수6329
- 분류기술동향
-
자료발간일
2012-05-18
-
출처
Riken Research
-
원문링크
-
키워드
#protein#hypothesis#neurodegeneration# pathogenic mechanism #prion diseases
Proving the protein-only hypothesis in neurodegeneration
Motomasa Tanaka at the RIKEN Brain Science Institute has made key advances in understanding protein-only infection and probing the pathogenic mechanism for prion diseases
Motomasa Tanaka
Team Leader
Laboratory for Protein Conformation Diseases
RIKEN Brain Science Institute
Creutzfeldt-Jakob disease, bovine spongiform encephalopathy (BSE) and other prion diseases are neurodegenerative conditions that develop due to abnormalities in a class of proteins known as prions. Unlike bacterial or viral infections, prion diseases have long been discussed by many researchers based on the protein-only hypothesis, in which prions are viewed as the only source of infection. Heading the Laboratory for Protein Conformation Diseases at the RIKEN Brain Science Institute, Motomasa Tanaka, and his colleagues verified the hypothesis for the first time in the world using yeast prions. Tanaka is analyzing the distinct mechanism by which a broad range of symptoms develop depending on protein aggregate (amyloid) structures. Using these results, Tanaka aims to clarify the pathogenesis of neurodegenerative diseases which have no established therapies, and to facilitate the development of new therapeutic treatments.
Prion diseases: a major public health concern
A prion disease is a debilitating condition characterized by neuron death in the brain, causing severe mental and physical deterioration and reducing brain tissue to a honeycombed, sponge-like appearance. If the illness develops after a long latency, symptoms such as cognitive impairment and delusions progress rapidly, leading to physical and mental deterioration within one to two years, eventually resulting in death. Well-known prion diseases include scrapie in sheep, bovine spongiform encephalopathy (BSE, or ‘mad cow disease’) in cattle, and Creutzfeldt-Jakob disease (CJD) in humans.
During the 1990s outbreak of CJD in the UK, evidence showed that cattle given feed which contained brain remnants from scrapie-infected sheep, meat and bone meal contracted BSE, and subsequently humans who consumed the meat of BSE-infected cattle later developed CJD. The risk that prion diseases could jump the species barrier sparked great public health concern at that time.
“Pathogens for infectious diseases are usually bacteria, viruses and the like, and any pathogen that has once infected a living organism can replicate nucleic acids (DNA and RNA) and proliferate autonomously,” explains Tanaka. “In prion diseases, however, neither bacteria nor viruses are the pathogens, with no nucleic acids detected in the body. Additionally, even within the same prion disease, different patients suffer damage to quite different areas of the brain, as well as different speeds of progression, symptoms and the like (Fig. 1). The mechanism behind the pathogenesis remains unclear, and no radical treatment is available.”
The protein-only hypothesis
In 1982, Dr Stanley Prusiner from the US discovered a fine substance in the brain of a patient with a neurodegenerative disease. The substance was identified as a protein consisting of 253 amino acids, and became known as a ‘prion’. Dr Prusiner was later awarded the 1997 Nobel Prize in Physiology or Medicine for his discovery of prions. “Dr Prusiner proposed the protein-only hypothesis that prion diseases are caused by infectious proteins called prions, rather than by bacteria and viruses. He hypothesized that in addition to normal prions, there are abnormal prions having exactly the same amino acid sequences as those of the normal type, and the etiology is attributed to abnormal prions (Fig. 1),” says Tanaka.
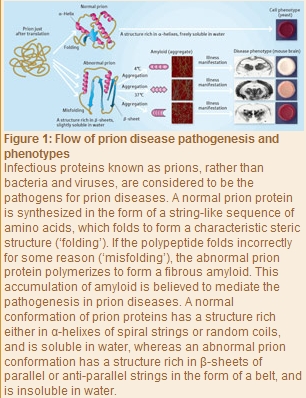
“Any protein can work normally provided its sequence of amino acids folds properly. This process is called folding. Abnormal prions are the result of misfolded amino acids, and have many band-like structures known as β -sheets. In the protein-only hypothesis, the structure of an abnormal prion is thought to act as a template for a normal prion, which is converted to the structure of the abnormal type, and in which fibrous amyloids form gradually.”
Although currently available experimental data support the protein-only hypothesis, it is controversial because the concept that infections can occur with proteins alone contradicts established medical dogma. “To verify this hypothesis, it must be demonstrated that prions are the only source of infection for prion diseases, with no other factors involved. It is also necessary to solve the riddle of why distinct phenotypes such as severity and diseased-site specificity emerge from prions having the same amino acid sequence,” says Tanaka.
Verification of the hypothesis
Tanaka and Jonathan Weissman (University of California, San Francisco) first attempted to show that prions are the sole source of infection for prion diseases in 2004, when they cultivated an amyloid for a yeast prion. “Yeast, which is a single-cell organism, has proteins that behave like mammalian prion protein, and these are called yeast prions. We created an amyloid for the Sup35 yeast prion and developed a technique for introducing it directly into uninfected yeast. The infection with Sup35 amyloid converted nearly 100% of the non-prion yeast to the prion state. The infection was established with the aggregated form of Sup35 alone.”
Tanaka next tried to unravel the puzzle of why distinct phenotypes emerge from prions having the same amino acid sequence by examining amyloid structures (Fig. 1). “We developed a method of generating amyloids with different ‘conformations’ (steric configurations of various atoms in a molecule) from Sup35 proteins which had the same amino acid sequence. The key point is that the Sup35 proteins polymerized merely at different temperatures in the absence of additives,” explains Tanaka. “We examined two amyloids polymerized at 4°C and 37°C respectively, and actually identified different conformations. We then conducted an infection experiment by introducing each of these two amyloids directly into yeast not infected with prion. As a result, two different colors developed: the yeast incorporating the amyloid polymerized at 4°C turned white, whereas the yeast incorporating the amyloid polymerized at 37°C turned pink. The infection rate also differed, with the amyloid polymerized at 4°C causing prion infections more efficiently than that at 37°C.”
More importantly, the phenotype (in this case, color) appearing in this experiment did not depend on the amount of amyloid introduced. “We used Sup35 proteins having the same amino acid sequence. Hence, we found that the generation of different phenotypes was caused by the difference in conformation, rather than the amount of amyloid.”
Amyloid structure’s role in Huntington’s disease
The diversity of amyloid structure was also found to feature in the pathogenesis not only of prion diseases, but also in the neurodegenerative condition of Huntington’s disease. An intractable disease characterized by involuntary movement, cognitive decline and psychiatric problems, Huntington’s disease has no known cure.
In the process of protein synthesis, bases are arranged in the gene domain of the DNA (base sequence) to represent ciphers for the arrangement of amino acids. A combination of only three of the bases adenine (A), thymine (T), guanine (G) and cytosine (C) defines one amino acid, and these amino acids join together to form a protein.
“In exon 1, which is one of the gene domains of huntingtin - the protein that causes Huntington’s disease - the CAG repeat is present and consists of a repeat sequence of the C, A and G combination,” explains Tanaka. “This repeat sequence defines the amino acid glutamine, and a long chain of glutamine units form polyglutamines. In patients with Huntington’s disease, more than 36 CAG repeats are found - which is more than the normal length of 7?35 - and they produce an abnormally elongated polyglutamine. The huntingtin containing this abnormal polyglutamine tends to form amyloid and plays a role in the pathogenesis of Huntington’s disease. However, the detailed pathogenetic mechanism remains unclear.”
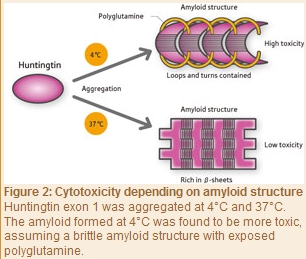
As with yeast prion, Tanaka and his colleagues in 2009 induced aggregation of the exon 1 of purified huntingtin at 4°C and 37°C to form amyloids. “The amyloid formed at 4°C was a brittle structure containing less β-sheets but more loops with a string-like structure and turns with a U-shaped loop structure, and with some of its polyglutamine units exposed to the outside of the aggregate (Fig. 2),” says Tanaka. “On the other hand, the amyloid aggregated at 37°C was a firm structure rich in β-sheets, in which its polyglutamine units were buried in the aggregate. Interestingly, an amyloid that was structurally very similar to the amyloids aggregated at 4°C and 37°C in vitro was observed in the brain of a mouse model of Huntington’s disease. We also found that the amyloid structure varied depending on the area in the brain. Furthermore, we conducted examinations to determine whether amyloid structural differences are associated with differences in the degree of toxicity, and found that the amyloid formed at 4°C was highly toxic, whereas that formed at 37°C was weakly toxic. These results correlate with our prion yeast findings. This is presumably because the exposed polyglutamines recruit normal proteins around them into the aggregates, resulting in loss of the function of normal proteins.”
Amyloid structuring process
“Many researchers, including myself, had a vague perception that highly infectious or highly toxic amyloids were in a hard set state,” explains Tanaka. “Of course, there was no reason for that. However, the reality was the opposite; the amyloids of the highly infectious yeast prion protein and highly toxic huntingtin were found to be rather brittle structures. Brittle amyloids are efficiently fragmented in cells. It is known that individual amyloid fragments act independently to increase infectivity. So the question arises as to how different amyloid structures are generated from proteins with the same amino acid sequence. In particular, elucidating the mechanism by which highly infectious or highly toxic amyloid structures are generated would help clarify the pathogenesis in neurodegenerative diseases caused by amyloids, such as prion diseases and Huntington’s disease, and accelerate the development of new therapeutic strategies.”
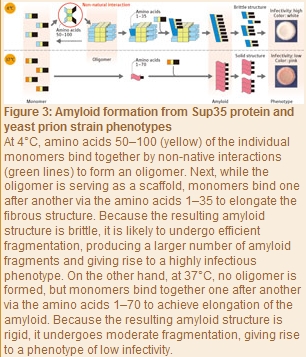
Later in 2010, Tanaka and his colleagues extensively examined the processes of amyloid formation at 4°C and 37°C with Sup35 proteins, using the SPring-8 syncrotoron radiation facility at the RIKEN Harima Institute. “We used a technique known as small-angle X-ray scattering by which a protein solution is exposed to X-rays, and the intensity of the resulting scattering is measured to determine the protein structure. As a result, we found Sup35 proteins to be monomers at 37°C, but at 4°C a stable oligomer was formed comprising many monomers bound together (Fig. 3). Also, even when the temperature was repeatedly d between 4°C and 37°C, the scattering intensity was constant at each temperature, so we found that the oligomer formation with a Sup35 protein is reversible through temperature . Furthermore, the amyloids formed at 4 to 9°C via oligomers were highly infectious and brittle structures, whereas the amyloids formed at 10 to 37°C without detectable oligomers were less infectious and rigid structures. In other words, whether a stable oligomer is formed proved to be the critical determinant of the eventual structure and phenotype of amyloids.”
The researchers next identified the amino acids involved in oligomer formation at 4 to 9°C by generating some Sup35 protein variants and using small-angle X-ray scattering to determine those amino acids which reduced the oligomer content. As a result, the first 100 amino acids in the prion domain (residues 1?123) proved to be involved in the oligomerization.
“We also found that oligomer formation was initiated when amino acids 50?100 in the prion domain bound together (Fig. 3),” says Tanaka. “According to a report by another study group, the ing 35 amino acids were identified as the amino acids used in the amyloid formation at 4°C, and the ing 70 amino acids at 37°C. These findings demonstrate that at 4°C monomers interact via amino acids 50?100 in each (non-native interaction) to initially form an oligomer. Subsequently, while the oligomer is serving as a scaffold, monomer units bind one after another to elongate the fibrous structure. Because the oligomer formation reduces the number of amino acids used in this elongation process to the first 35 residues in the prion domain, a brittle amyloid structure is eventually produced. By contrast, at 37°C - where no oligomer is formed - the initial 70 amino acids in the prion domain can take part in the formation of an amyloid core, a level which is double at 4°C, so a stable rigid amyloid structure forms.” Brittle amyloids are likely to undergo efficient fragmentation in cells, resulting in the generation of more amyloid fragments and increased infectivity.
Amyloid structuring process
“Many researchers, including myself, had a vague perception that highly infectious or highly toxic amyloids were in a hard set state,” explains Tanaka. “Of course, there was no reason for that. However, the reality was the opposite; the amyloids of the highly infectious yeast prion protein and highly toxic huntingtin were found to be rather brittle structures. Brittle amyloids are efficiently fragmented in cells. It is known that individual amyloid fragments act independently to increase infectivity. So the question arises as to how different amyloid structures are generated from proteins with the same amino acid sequence. In particular, elucidating the mechanism by which highly infectious or highly toxic amyloid structures are generated would help clarify the pathogenesis in neurodegenerative diseases caused by amyloids, such as prion diseases and Huntington’s disease, and accelerate the development of new therapeutic strategies.”
Later in 2010, Tanaka and his colleagues extensively examined the processes of amyloid formation at 4°C and 37°C with Sup35 proteins, using the SPring-8 syncrotoron radiation facility at the RIKEN Harima Institute. “We used a technique known as small-angle X-ray scattering by which a protein solution is exposed to X-rays, and the intensity of the resulting scattering is measured to determine the protein structure. As a result, we found Sup35 proteins to be monomers at 37°C, but at 4°C a stable oligomer was formed comprising many monomers bound together (Fig. 3). Also, even when the temperature was repeatedly d between 4°C and 37°C, the scattering intensity was constant at each temperature, so we found that the oligomer formation with a Sup35 protein is reversible through temperature . Furthermore, the amyloids formed at 4 to 9°C via oligomers were highly infectious and brittle structures, whereas the amyloids formed at 10 to 37°C without detectable oligomers were less infectious and rigid structures. In other words, whether a stable oligomer is formed proved to be the critical determinant of the eventual structure and phenotype of amyloids.”
The researchers next identified the amino acids involved in oligomer formation at 4 to 9°C by generating some Sup35 protein variants and using small-angle X-ray scattering to determine those amino acids which reduced the oligomer content. As a result, the first 100 amino acids in the prion domain (residues 1?123) proved to be involved in the oligomerization.
“We also found that oligomer formation was initiated when amino acids 50?100 in the prion domain bound together (Fig. 3),” says Tanaka. “According to a report by another study group, the ing 35 amino acids were identified as the amino acids used in the amyloid formation at 4°C, and the ing 70 amino acids at 37°C. These findings demonstrate that at 4°C monomers interact via amino acids 50?100 in each (non-native interaction) to initially form an oligomer. Subsequently, while the oligomer is serving as a scaffold, monomer units bind one after another to elongate the fibrous structure. Because the oligomer formation reduces the number of amino acids used in this elongation process to the first 35 residues in the prion domain, a brittle amyloid structure is eventually produced. By contrast, at 37°C - where no oligomer is formed - the initial 70 amino acids in the prion domain can take part in the formation of an amyloid core, a level which is double at 4°C, so a stable rigid amyloid structure forms.” Brittle amyloids are likely to undergo efficient fragmentation in cells, resulting in the generation of more amyloid fragments and increased infectivity.
Switching by protein
“Although we previously viewed only amyloids as the cause of diseases, we have recently come to think that they may play roles in controlling cell functions in the body,” explains Tanaka. “A soluble state of protein (the normal type in prion diseases) and an aggregated state of protein (the abnormal type in prion diseases) are in fact relatively common phenomena; for example, they may inter with each other according to environmental s. For example, the soluble type works to switch off a function, whereas the aggregated type works to switch it on, and they can both adapt to environmental s. If the same switching is to be achieved by genomic s, there may arise the risk of causing a mutation in some site; however, if switching is possible with proteins alone, the risk is avoidable,” says Tanaka, who has ed new research aiming to discover new cell functions based on reversible switching of the soluble and aggregated states of proteins such as prions.
“Of course, we will also conduct research that will illuminate the pathogenesis in neurodegenerative diseases such as prion diseases and Huntington’s disease, and lead to the development of new therapeutic strategies and drug discoveries.” In a yeast prion, a fragmenting protein that plays a key role in increasing infectivity has already been identified. It has been established that, if this fragmenting protein is functionally inhibited, the amyloid continues to elongate without undergoing fragmentation and then returns from the prionized state to the normal state. Applying this mechanism may lead to a new treatment which blocks the fragmenting factor. “However, many problems remain to be solved. For example, no fragmenting protein has been discovered in mammals, and none have succeeded in achieving efficient prion infections using protein alone. In the case of mammals, some researchers suggest that highly infectious amyloids do not form without auxiliary factors such as nucleic acids. We are conducting research to open up new routes, taking advantage of the discovered mechanism in yeast prions.”
Motomasa Tanaka
-
다음글
- 다음글이 없습니다.
-
이전글
- 이전글이 없습니다.
지식
동향
- 기술동향 표적 단백질 분해 기술(TPD, Targeted protein degradation) 및 그 파생기술 연구 동향 2022-12-08
- 기술동향 PROTACs: Reinvention of an old workhorse-the small molecule drug 2019-12-11
- 기술동향 SMC proteins 2017: Chromosomal organizers from bacteria to human 참관 후기 2017-09-19
- 기술동향 System biology 동향: Protein-protein interaction network 구축현황과 활용 2017-03-29
- 기술동향 단백질 칩의 응용과 활용전망 2014-01-08