

산업동향
Review of FDA CDER Approved Drugs in 2019(3)
- 등록일2020-05-26
- 조회수4907
- 분류산업동향 > 제품 > 바이오의약
-
자료발간일
2020-04-27
-
출처
(재)범부처신약개발사업단
- 원문링크
-
키워드
#FDA#CDER
|
Drug Name |
Indication(s) |
Pharmacologic Class/Unmet Medical Needs |
|
Nourianz (Istradefylline) |
Nourianz is indicated as adjunctive treatment to levodopa/carbidopa in adult patients with Parkinson’s disease (PD) experiencing ‘off’ episodes.
Patients enrolled were on a stable levodopa/dopa decarboxylase inhibitor (DCI) regimen.
*Off episodes or off time refer to periods in which patients have greater difficulty with motor symptoms and includes issues with mobility, slowness, and stiffness. |
Nourianz is an adenosine A2A receptor antagonist. It is a structural analog of caffeine and a derivative of xanthine.
In animals, nourianz has been shown to potentiate and prolong the effects of levodopa in experimental animal models of Parkinson’s disease. The sponsor hypothesizes that blockage of the A2A receptors by nourianz reduces the excitability of this indirect pathway of the basal ganglia, resulting in an improvement in Parkinson’s disease symptoms without producing abuse potential or physical dependence.
The efficacy of nourianz for the adjunctive treatment to levodopa/carbidopa in patients with
Parkinson’s disease experiencing “off” episodes was shown in four randomized, multicenter, double-blind,
12-week, placebo-controlled studies (Study 1, NCT00456586; Study 2, NCT00199407; Study 3,
NCT00455507; and Study 4, NCT00955526). The studies enrolled patients with a mean duration of
Parkinson’s disease of 9 years (range: 1 month to 37 years) that were Hoehn and Yahr Stage II to IV,
experiencing at least 2 hours (mean approximately 6 hours) of “off” time per day, and were treated
with levodopa for at least one year, with stable dosage for at least 4 weeks before screening (mean total daily dosage range: 416 to 785 mg).
The primary efficacy endpoint was the change from baseline in the daily awake percentage of “off”
time, or the change from baseline in total daily “off” time, based on 24-hour diaries completed by
patients. A change from baseline in “on” time without troublesome dyskinesia (i.e., “on” time without dyskinesia plus “on” time with non-troublesome dyskinesia) was a secondary efficacy endpoint.
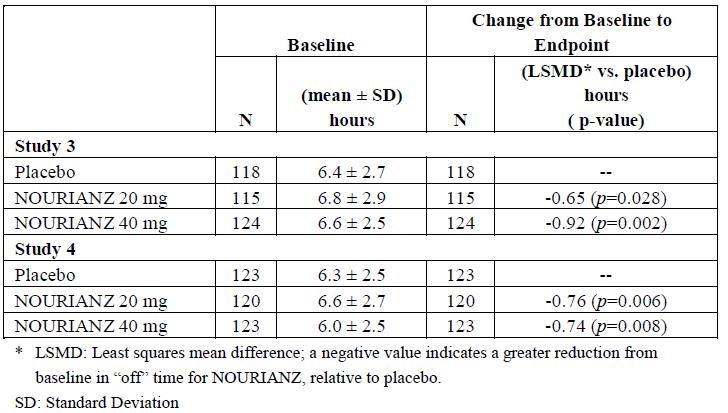
In Study 1 and 2, change from baseline in daily awake off time of placebo vs nourianz was 37.2 ± 13.8 and 38.4 ± 16.2, respectively in Study 1. In Study 2, 38.7 ± 11.6 and 39.8 ± 14.0, respectively. Compared with patients on placebo, patients treated with nourianz experienced an additional increase from baseline in “on” time without dyskinesia of 0.96 hours (nominal p=0.025) in study 1, and of 0.55 hours (nominal p=0.315). The results of Study 3 and 4 was attached as table.
Table: Change from Baseline in Daily OFF time
Good safety profile and improvement in efficacy as an adjuctive treatment enables FDA approval. |
|
Oxbryta (vexelotor) |
Oxbryta is indicated for the treatment of sickle cell disease (SCD) in adults and pediatric patients 12 years of age and older. This indication is approved under accelerated approval based on increase in hemoglobin (Hb). Thus further verification and description of clinical benefit in confirmatory trial(s) are required for full approval. |
Oxbryta is hemoglobin S polymerization inhibitor that binds to HbS with a 1:1 stoichiometry and exhibits preferential partitioning to red blood cells (RBCs). By increasing the affinity of Hb for oxygen, oxbyta demonstrate dose-dependent inhibition of HbS polymerization. Nonclinical studies suggest that oxbryta may inhibit RBC sickling, improve RBC deformability, and reduce whole blood viscosity.
The pharmacodynamics effect of oxbryta treatment demonstrated a dose-dependent increase of Hb oxygen affinity as determined by the change in p50 (partial pressure of oxygen at which Hb oxygen saturation of 50% is achieved) that was linearly correlated with oxbryta exposure. Oxbryta demonstrated a dose-dependent reduction in clinical measures of hemolysis (indirect bilirubin and % reticulocytes). HOPE trial [INCT03036813]. The overall safety profile of oxybryta appears acceptable for proposed registrational dose of 1,500mg and support a favorable benefit-risk assessment for oxbryta for patients with SCD.
In addition, the background of the accelerated approval is that the treatment of oxbryta demonstrate a decrease in the risk of strokes for patients with SCD since patients with SCD experience significant morbidity due to the risk of strokes including silent strokes. |
|
Padcev (enfortumab vedotin-ejdf) |
Padcev is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer who have previously received a programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor, and a platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally advanced or metastatic setting.
Patient population is metastatic UC patient with FGFR3 or FGFR2 genetic alterations in the post-platinum setting; however, only approximately 20% of patients with metastatic US have these FGFR alterations. This indication is also approved under accelerated approval based on tumor response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
There are no other approved therapies for the proposed patient population. |
Padcev is a Nectin-4-directed antibody-drug conjugate.it is comprised of a fully human IgG1-kappa antibody (AGS-22C3) conjugate to the microtubule-disrupting agent, monomethyl auristatin E, via a protease-cleavable maleimidocaproyl valine-citrulline linker (SDG-1006). Approval of padcev is the first approval of a drug that specifically targets the Nectin-4 pathway.
The efficacy of padcev was evaluated in EV-201[NCT03219333], single-arm, multicenter trial that enrolled 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy.
Patients were excluded if they had active CNS metastases, ongoing sensor or motor neuropathy≥Grade 2, or uncontrolled diabetes defined as hemoglobin A1c (HbA1c)≥8% or HbA1c≥7% with associated diabetes symptoms.
The major efficacy outcome measures were confirmed objective response rate (ORR) and duration of response (DOR) assessed by blinded independent central review (BICR) using RECISTv1.1.
The confirmed objective response rate (ORR) in 125-patient efficacy population was 44% (95% CI) and the median duration of response (DoR) was 7.6 months (95% CI) as determined by the Blinded Independent Central Review (BICR). The median follow-up duration was 10.2 months. In post-platinum setting, trials report ORR less than 15% for single agent therapy.
Approval of padceb us recommended by all disciplines. Results of the ongoing randomized phase 3 trial EV-301
will serve as a PMR to confirm the clinical benefit of padceb. |
|
Polivy (polatuzumab vedotin-piiq) |
Polivy is indicated for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) in combination with bendamustine and a rituximab product, not otherwise specified, after at least two prior therapies.
Accelerated approval was granted for this indication based on complete response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. |
Polivy is a CD79b-directed conjugate with activity against dividing B cells. The small molecule, MMAE is an anti-mitotic agent covalently attached to the antibody via a cleavable linker. The monoclonal antibody binds to CD79b, a B-cell specific surface protein, which is a component of the B-cell receptor. Upon binding CD79b, polivy is internalized, and the linker is cleaved by lysosomal protease to enable intracellular delivery of MMAD. MMAE binds to microtubules and kills dividing cells by inhibiting cell division and inducing apoptosis.
Patients with relapsed or refractory DLBCL have unmet medical need. Although axicabtagene ciloleucel and tisagenlecleucel are approved for R/R large B-cell lymphoma, these therapies are not widely available, and have specific requirements for therapy.
The efficacy and safety results from clinical trial GO29365(NCT02257567) demonstrate substantial evidence of effectiveness and an acceptable benefit-risk profile for polivy, in combination with bendamustine and rituximab product, for the treatment of adult patients with relapsed or refractory disuse large B-cell lymphoma.
The efficacy of polatuzumab vedotin-piiq (pola) is based on complete response (CR) rate assessed by an independent review committee (IRC), best overall response rate (BOR), and duration of response (DOR) in Study GO29365, which included a randomized, open-label, cohort of 80 patients with relapsed or refractory DLBCL.
Patients were randomized 1:1 to receive either pola + bendamustine and rituximab (BR) or BR
alone for six 21-day cycles. Eligible patients were not candidates for autologous hematopoietic stem cell transplantation (HSCT) at study entry.
The primary endpoint was IRC-assessed CR rate by PET-based criteria at end-of-treatment (EOT). The median number of prior therapies was 2, with 29% receiving one prior therapy, 25% receiving 2 prior therapies, and 46% receiving 3 or more prior therapies. Eighty percent of patients had refractory disease to last therapy.
Patients in the pola +BR arm received a median of 5 cycles, whereas patients in the BR arm received a median of 3 cycles. The difference in
exposure was driven by the higher rate of early treatment discontinuation due to inefficacy in the BR arm. At EOT, the FDG-PET CR rate per
IRC was 40% with pola + BR vs. 18% with BR. This represented a 22% higher observed CR rate (95% CI: 3, 41) in the pola arm. A best response of CR or PR per IRC was achieved by 63% of the pola + BR arm and 25% of the BR arm, whereas a best response of CR was achieved by 50% and 23%, respectively.
Responses tended to be more durable in the pola + BR arm. Of the 25 responding patients in the pola + BR arm, 16 (64%) had a DOR of ≥ 6 months, and 12 (48%) had a DOR of ≥ 12 months. Of the 10 responding patients in the BR arm, 3 had a DOR lasting ≥ 6 months, and 2 had a DOR lasting ≥ 12 months. Although progression-free survival (PFS) and overall survival (OS) were available in Study GO29365, the small number of events and sample size limit the interpretability of the PFS and OS results.
Based on CR rate at EOT, BOR, and DOR per IRC, there is a strong indication of a positive treatment effect with the addition of pola to BR in the intended population. In addition to positive treatment effect, its acceptable safety profile enables approval. |
☞ 자세한 내용은 내용바로가기 또는 첨부파일을 이용하시기 바랍니다.
지식
동향