

기술동향
Single cell genomics 연구 및 기술 동향
- 등록일2015-11-10
- 조회수9243
- 분류기술동향
-
자료발간일
2015-11-10
-
출처
생물학연구정보센터(BRIC)
- 원문링크
-
키워드
#단일세포#genomic#기술동향#유전체학#Single cell
- 첨부파일
 pdf_0002421.pdf
pdf_0002421.pdf
출처 : 생물학연구정보센터(BRIC)
Single cell genomics 연구 및 기술 동향
저자 : 노규형 (가톨릭대학교)
1. 서론
단일세포 생물학과 유전체 생물학은 서로 가까워져서 지금 만나고 있는 중이다. 특정한 유전자의 number를 측정하는 방법으로 임상에서 많이 쓰이는 FISH 의 결과값은 비교적 큰 값인데, 예를 들어 ERBB2의 경우 ASCO/CAP 가이드에 따르면 6 이상인 경우에 대해 ERBB2 양성(positive)으로 판정한다 [Wolff 2013]. 반면 차세대 시퀀싱(NGS) 데이터로부터 계산한 암 조직의 number 값이 3을 초과하는 것을 보기는 어렵다. 이것은 FISH는 현미경으로 형광 신호를 보이는 암세포들에 대해서 분석하는데 반해, 시퀀싱 데이터를 생산하기 위해 준비하는 시료는 개별 세포가 아닌 덩어리이며, 이 덩어리의 불균일성 (heterogeneity) - 채취된 조직 안에 정상세포가 상당량 포함되어 있을 뿐 아니라, 암세포간의 유전형 또한 불균일하다 - 이 number 값의 평균을 낮추기 때문이다. 불균일성은 시료에만 있는 것은 아니고 세포 수준에서도 존재하는데, 단세포 생물에서 고등생물에 이르기까지 세포 간의 유전형, 표현형의 차이가 있다는 것은 잘 알려진 사실이다. 모자이시즘 (mosaicism)은 동일한 개체를 이루는 세포들이 서로 다른 유전형을 가질 수 있다는 예를, 줄기세포에서 분화된 각 세포는 동일한 유전형을 가지지만 표현형에 있어서는 차이가 있는 경우를 보여주는 예라고 할 수 있겠다.
이러한 불균일성과 그 결과를 연구하려면 지금까지는 FISH와 같은 소량 데이터 생산 기술을 이용하여 단일세포 내에서 제한된 수의 측정값을 얻는 수밖에 없었다. 한편 유전체 기술의 발달로 고속 대량 데이터의 생산과 분석이 이루어지고는 있으나 수천 혹은 수백만 개 세포로부터 얻어진 앙상블 평균에 기반하고 있다는 점에서 세포 수준에서의 다양성에 대한 연구 가능성은 처음부터 배제된 셈이다. 그러나 이제 시퀀싱 기술과 분자생물학의 발전으로 단일세포의 유전체 연구가 가능하게 되면서 유전학과 단일세포 생물학의 융합이 일어나고 있다. (그림1)
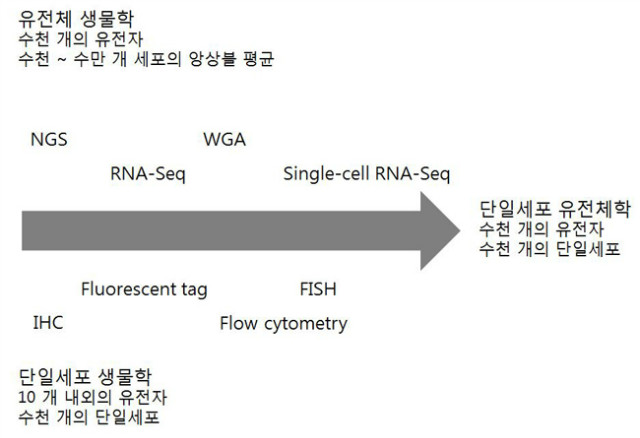
그림 1. 단일세포 생물학과 유전체 생물학의 융합
그리고 초기의 기술적인 이슈였던, 단일 세포의 DNA 시퀀싱에 어떤 증폭 방법을 써야 할 것인가 고민하던 단계에서, 단일 세포의 RNA 시퀀싱 [Macosko 2015]을 넘어 이제는 microdroplet을 이용하여 대량의 자동화된 시퀀싱에 대한 연구로 진행되어 가고 있다. 단일세포 시퀀싱에 대한 전반적인 리뷰는 [Baslan 2014, Kolodziejczyk 2015, Liang 2014, Shapiro 2014]을 읽어보기 바란다.
2. 단일세포 유전체학의 분석 수행 과정
Capture and lysis -> ion -> pre-amplification -> sequecning 의 순서로 진행된다. 단일세포를 채취하는 과정과 cell lysis를 통하여 필요한 유전물질을 획득하는 과정이 우선 필요하다. DNA를 연구하기 위해서는 lysis 만으로 충분하나, mRNA를 연구하려면 poly-A tail을 이용한 capturing이 필요하다. 또한 세포 하나에서 얻을 수 있는 DNA의 양은 picogram정도이므로 시퀀싱이 가능한 수십 ~ 수백 nanogram 수준으로 충분히 늘리는 증폭 과정 (pre-amplification)이 필요하다.
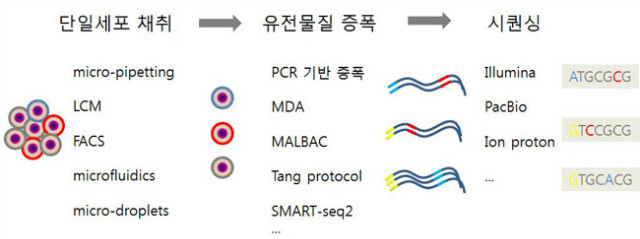
그림 2. 단일세포 유전체학의 분석 수행 과정 (single cell omics procedure)
3. 단일세포의 채취 (Single-cell capture)
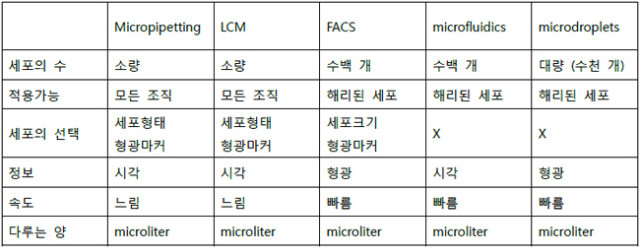
표1. 단일세포의 채취방법
가. micropipetting
우리에게는 난자 세포를 다루는 기술로 많이 알려진 micropipetting은 현미경으로 보면서 마이크로 피펫을 이용해서 분리해내는 방법으로 배아세포 등을 분리하는데 많이 사용되고 있으나 시간이 많이 걸리므로 대량의 세포를 채취하기에는 무리가 있다 [Grindberg 2013].
나. laser capture
여러 가지 세포가 구성된 조직에서 원하는 세포를 분리 채취하는 방법으로 Laser Capture Microdissection (LCM)이 사용된다. Thermoplastic transfer film이 얇게 입혀진 LCM 전용 cap을 조직 절편 위에 올려두고 현미경으로 보면서 원하는 세포부위에 저출력의 레이저를 쪼이면 transfer film이 녹아서 원하는 세포와 붙는데, 나중에 cap을 분리하면 나머지 조직은 슬라이드에 그대로 있고, transfer film에 의해 붙은 세포들만 분리해낼 수 있다 [Frumkin 2008]. 적출한 암조직에는 정상세포와 암세포가 섞여 있는데 이를 분리하는 방법으로 많이 사용된다.
위의 두 방법은 수동으로 단일세포를 분리해야 하므로 수율이 매우 낮은데, 조직을 trypsin 혹은 collagenase 등으로 처리하여 해리 (dissociation)시킨 단일세포 현탁액 (cell suspension)을 만들면 자동화하기 쉬운 상태가 되어 FACS나 microfluidics 장비, microdroplet 장비를 이용할 수 있다. 물론 이러한 과정이 세포 활성이나 전사체 발현 패턴에 영향을 줄 수 있다는 점은 고려해야 한다.
다. Fluorescence-activated cell sorting (FACS)
FACS는 각 세포에서 검출되는 형광의 세기나 cell size에 따라 자동으로 세포를 분류해주는 장치이다. 세포 현탁액은 FACS를 통과하면서 작은 방울 형태가 되는데 각각 하나의 세포가 들어가도록 조정되며, 검출된 정보에 따라 레이저를 쬐어 정전기 전하를 가지도록 한다. 그 아래에 위치한 정전기 편향 시스템을 지날 때 이 정전기 값에 따라 궤도가 바뀌므로 서로 다른 용기로 분리할 수 있게 된다 [Hayashi 2010].
라. microfluidics
대표적인 장비인 Fluidigm C1은 각 chip의 96개 well에 각각 세포 하나씩을 자동으로 분리해준다. 제약 조건은 세포가 구형이며, 크기가 동일해야 한다는 것이다. 구형이 아닌 세포의 경우는 효율이 매우 떨어진다고 한다. 또한 한 번의 시행에 천 여개 이상의 세포가 필요하다고 한다. 이렇게 분리된 각 단일 세포에 대해 역전사 및 증폭 등 시퀀싱에 필요한 실험 자동화가 가능하다 [Shalek 2013].
마. microdroplets
현재 가장 주목받는 최신 기술인데, 세포들이 microdroplet에 하나씩 들어가도록 조정하고, 이렇게 분리된 microdroplet 내에서 실험 반응이 일어나도록 하는 기술로 수천 개의 단일세포를 단시간에 처리할 수 있다 [Mazutis 2013]. 대표적인 논문 [Klein 2015] 에서 사용된 사례를 보면 각각 서로 다른 UMI (unique molecular identifier)로 태깅된 primer가 들어있는 hydrogel과 세포를 섞고 lysis buffer를 넣은 후 각 drop마다 세포와 hydrogel이 하나씩 들어가도록 한다. RNA-Seq을 진행하기 위해 reverse tranion을 수행하고 cDNA로 만든 후, droplet을 깨고 합쳐서 시퀀싱하고 얻어진 시퀀스를 UMI에 따라 분리, 분석 처리한다. UMI의 길이는 8bp로 이론상 48=65,536 개의 세포에 각각 고유한 인덱스를 붙일 수 있다. (그림 3)
그림 3. microdroplet
Klein 2015 (Cell 161, 1187?1201) Fig.1 A Platform for DNA Barcoding Thousands of Cells
(http://www.sciencedirect.com/science/article/pii/S0092867415005000#fig1)
4. Pre-amplification (WGA for single-cell DNA-Seq)
각 리뷰 논문에도 나와 있기는 하지만 각 전장유전체 증폭 방법에 대한 수치적인 비교는 Stanford 대학, Quake 그룹의 논문 [de Bourcy 2014]을 참고하면 좋겠다.
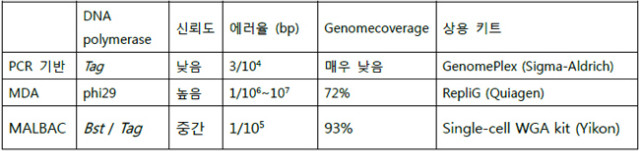
표2. 전장유전체 증폭 (WGA) 방법 비교
가. PCR 기반의 전장유전체 증폭 (WGA)
NGS 이전에 개발되었던 PCR 기반의 WGA들은 단일세포의 특정위치의 CNV, SNV를 보기 위해 개발되었던 것이었다. 잘 알려진 바와 같이 PCR은 증폭 대상 시퀀스에 대한 primer를 넣고 Tag DNA polymerase를 써서 denaturation, annealing, elongation을 반복함으로써 매 싸이클마다 두 배의 산출물을 얻는 기하급수적 증폭 방법이며, 증폭되는 DNA 절편의 크기는 0.1 ~ 10kb 이고 증폭 과정 중 rate은 104 base 마다 3개 정도이다. 특정 시퀀스가 아닌 전장유전체 증폭 (Whole genome amplification)을 하기 위해서는 여러 가지 방법이 사용되는데, LA-PCR (ligation anchored PCR)에서는 DNA를 적절하게 조각내고 adaptor를 붙이고 PCR하는 방식을 쓰는 반면 [Troutt 1992], PEP-PCR (primer extension pre-amplification PCR) 에서는 random primer를 써서 DNA의 여러 위치에서 증폭이 일어날 수 있도록 한다 [Zhang 1992]. DOP-PCR (degenerate oligonucleotide priming PCR) 의 경우 특정 위치의 시퀀스가 여러 염기가 되도록 가능한 모든 조합을 섞어 만든 degenerate primer를 DNA에 붙이면 primer와 유사한 시퀀스가 있는 여러 위치에 결합할 수 있게 되어 dense priming이 가능하게 된다고 한다 [Telenius 1992]. 두 단계의 PCR 과정이 있는데 첫 번째는 DNA template에서 PCR을 하고, 두 번째는 증폭된 시퀀스를 대상으로 PCR을 진행한다. 짧은 amplicon 길이에 기인한 증폭 바이어스가 있으나 60kb 정도의 number를 충실하게 재현하며 [Baslan 2012] 전반적인 genome coverage는 매우 낮다고 알려져 있다.
나. MDA
Multiple displacement amplification (MDA)는 6개의 nucleotide로 이루어진 random hexamer primer를 DNA 위에 붙인 후 phi29 DNA polymerase를 써서 일정한 온도에서 이루어지는 증폭 과정이다 [Dean 2002]. Elongation 도중 다른 DNA polymerase가 만들고 있는 strand를 만나면 strand displacement 가 일어나 곁가지 형태의 branch를 만들게 되고, 이 branch에 DNA polymerase가 다시 붙는 방식으로 증폭이 일어나므로 PCR처럼 denaturation, annealing 과정이 필요 없이 일정한 온도에서 증폭이 일어나게 된다. DNA polymerase가 오래 붙어있으므로 amplicon의 길이는 12kb 이상으로 길어지며, 에러율은 106 ~ 107 base 당 하나로 상당히 낮다. Genome coverage는 25x 대비 1x에서 72% 로 높은 편이다.
다. MALBAC
가장 최근에 개발된 multiple annealing and looping-based amplification cycles (MALBAC)은 MDA와 PCR을 결합한 형태로 degenerate primer를 DNA에 붙이고 Bst DNA polymerase를 써서 MDA를 진행하다가 denaturation 하게 되면 primer가 붙은 displaced DNA fragment가 떨어져 나오게 된다. 원 DNA와 primer가 붙은 displaced DNA fragment에 다시 primer를 붙이고 합성을 시작한 후 다시 denaturation 하면 원 DNA에서는 앞서와 같이 한 끝에만 primer가 붙은 displaced DNA fragment를 만들게 되지만, displaced DNA fragment 위에서 합성된 DNA는 양 끝에 primer가 붙어 있어 (full amplicon), hairpin loop을 형성하고 더 이상 pre-amplification이 일어나지 않는다 [Zong 2012]. 이 과정은 PCR처럼 denaturation, annealing, elongation 과정을 반복 진행하지만, hairpin loop의 형태를 하고 있는 full amplicon은 복제의 template으로서는 배제되므로 준선형적 증폭(quasi-linear amplification)이며 생성되는 full amplicon은 원본의 2차 복제본만으로 구성된다. 이 과정은 PCR의 기하급수적 증폭 (exponential amplification) 특성이 가진, 이전 단계의 복사본을 다음 단계의 증폭의 template으로 사용하면서 복제할 때 생길 수 있는 에러의 누적을 막을 수 있어 에러율을 다소 낮출 수 있게 되었다. 5회 반복 후 생성된 산물 (full amplicon)을 PCR하여 충분한 양을 확보한다. Genome coverage는 25x 대비 1x에서 93% 로 상당히 높다. Allele dropout이 있기는 하나 MDA가 원래의 10% 재현에 그친다면 MALBAC은 72% 정도 재현해낸다고 한다. 최적화 해야 할 부분이 남아있기는 하지만, 현재로서는 단일세포 유전체 연구에는 MALBAC이 가장 적절한 증폭방법으로 여겨진다 [Chi 2013, Lasken 2013]. 각 방법에 대한 다이어그램과 비교표를 참고하기 바란다. (그림4 / 표2)
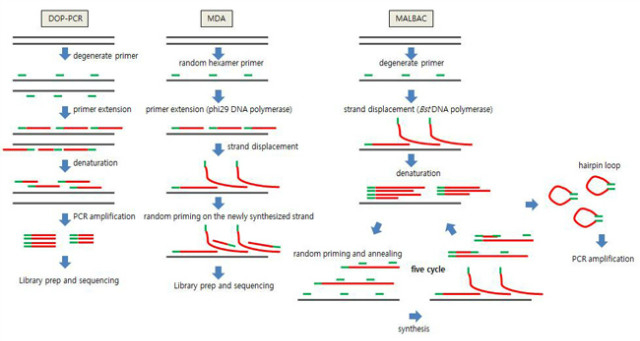
그림 4. 전장유전체 증폭 (WGA) 방법들
5. Pre-amplification (WTA for single-cell RNA-Seq)
전장발현체 증폭 방법에 대한 리뷰는 [Kolodziejczyk 2015, Liang 2014]을 참고하면 좋겠다.
표3. 전장발현체 증폭 (WTA) 방법 비교
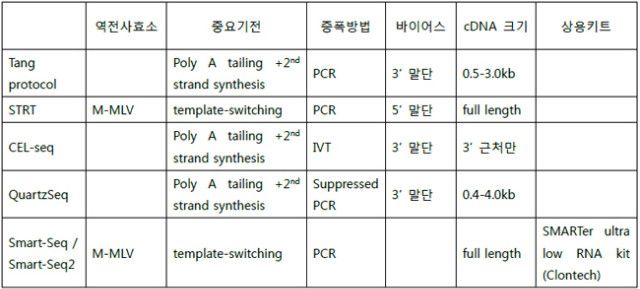
가. Tang protocol
최초의 단일세포 전사체 시퀀싱은 Poly-A tailing을 토대로 한 PCR 프토토콜로 [Tang 2009], 우선 단일세포를 lysis 한 후 anchor 시퀀스 (UP1)이 달린 poly(T) primer를 붙이고 역전사하여 cDNA를 만든 후 3’ 말단 부분에 poly(A) tailing을 해준다. 또 다른 anchor 시퀀스 (UP2)가 붙은 poly(T) primer를 이 cDNA에 붙이고 두 번째 cDNA를 합성하면 양 끝에 anchor 시퀀스 (UP1, UP2)가 붙은 cDNA를 만들 수 있게 되어서, 각 anchor 시퀀스를 primer로 PCR을 진행할 수 있게 된다. 이 방법은 3’ 말단에 치우치는 바이어스가 있는데, 이것은 주로 역전사 과정에서 역전사 효소가 mRNA의 끝까지 주행하지 못하면서 생기는 짧은 cDNA로 인한 것이다.
나. STRT
Tang protocol과 마찬가지로 처음의 cDNA 합성은 poly(T) primer로 시작하는데, 여기서는 M-MLV 역전사효소를 쓴다. 역전사를 하다가 5’ 말단에 이르면 역전사 효소의 terminal transferase 기능이 합성된 cDNA 끝 부분에 C 염기 몇 개를 붙여주게 되는데, 이 때 역전사 효소가 처음에 넣어준 G 몇 개가 붙은 시퀀스 조각으로 템플릿 갈아타기 (template switching)를 하여 fusion 된 cDNA 가 만들어지게 된다. 이후 여러 샘플을 pooling 하고 나중에 붙여준 시퀀스를 primer 로 하여 PCR을 수행한다 [Islam 2011]. 5’ 말단에서의 bead capture와 절편화 때문에 5‘ 말단에 치우치는 바이어스가 있다.
다. Cel-seq
Cel-seq에서는 Tang protocol을 따라 생성한 첫 번째, 두번째 cDNA를 in vitro tranion (IVT)를 이용하여 증폭한다. 다만 primer에 unique barcode를 붙이는 것이 다르다. IVT는 선형적 증폭 성능을 가지기 때문에 이론적으로는 PCR 기반의 증폭 과정에서 생기는 바이어스를 극복할 수 있다. [Hashimshony 2012].
라. Quartz-seq
Tang protocol처럼 처음의 cDNA 합성은 anchor 시퀀스가 붙은 poly(T) primer로 시작하고 5’ 말단에서 poly(A) tailing을 해 주는데, 두 번째 cDNA를 합성할 때 앞의 anchor 시퀀스가 붙은 poly(T) primer로 진행한다. 결과로 얻어진 cDNA 양 끝에 동일한 anchor primer가 hairpin loop을 만들어 주므로, 이후의 PCR 과정에서는 첫 번째 cDNA 만을 증폭하게 하는 효과가 있다 (suppressed PCR [Siebert 1995]). [Sasagawa 2013]
마. Smart-Seq / Smart-Seq2
STRT와 유사하게 진행하나 다른 점은 fusion된 cDNA를 anchor 시퀀스 및 fusion된 시퀀스를 primer 로 PCR을 한다는 것인데, 결과적으로 mRNA 일부가 아닌 전체를 대상으로 합성한 cDNA 만을 증폭하게 되어 3’ 바이어스를 줄일 수 있게 된다 [Ramskold 2012]. 2013년에 성능이 개선된 Smart-Seq2 이 나왔다 [Picelli 2013].
그림 5. 전장발현체 증폭 (WTA) 방법
Liang 2014 (Journal of Genetics and Genomics 41, 513-528) Fig. 3. Illustrative diagram of single-cell tranomic sequencing technologies에서 선택 발췌한 그림
(http://www.sciencedirect.com/science/article/pii/S1673852714001684#fig3)
6. 활용분야
가. Microbial ecology (metagenomics)
대부분의 미생물은 배양이 불가능하므로 유전자 마커를 이용하여 간접적으로 알아내는 방법을 쓴다. 군유전체학 (meta-genomics)에서는 특정 환경에 존재하는 미생물 군집에서 나오는 모든 유전체를 분석 대상으로 하는데, 이렇게 혼합된 유전체를 시퀀싱하고 데이터베이스와 비교하여 어떤 미생물이 있는지 알아내는 연구를 한다 [Handelsman 2004]. 종 다양성 분석과 계통 분석을 위해서는 보통 16S rRNA 부분만을 시퀀싱하여 분석하지만 유전자 예측을 하기 위해서는 전장 메타지놈 (Whole metagenome) 시퀀싱을 수행한다. 이렇게 예측된 유전자는 어떤 미생물에서 온 것인지 알 수 없었으나, 단일세포 시퀀싱이 가능해지면서 특정 유전자와 미생물 간의 관계를 직접적으로 알 수 있게 되었다 [Rinke 2013]. 단일세포 시퀀싱은 미생물 연구 분야에서 그동안 발견하기 어려웠던 아종을 밝히고 더 깊게 이해할 수 있도록 해줄 것이다. [Lasken 2015]
나. Haplotypes and somatic variation
개체의 세포 종류에 따라 서로 다른 유전형, 표현형을 보려는 시도는 이미 진행되고 있었다. Sperm cell 의 유전자재조합 비율 및 패턴을 측정하는 연구 [Lu 2012], 앞서 예로 들었던 모자이시즘 연구 [McConnell 2013], 배아 발달 단계에서의 염색체 불안정성 연구 [Vanneste 2009], 혈액 내 각 세포의 발현 패턴에 대한 연구 [Macosko 2015] 등이 있다.
다. Cancer research
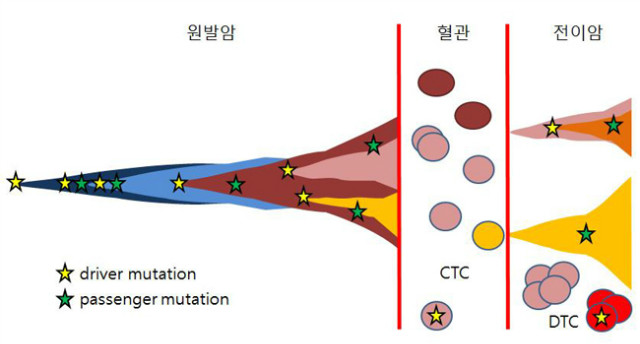
그림 6. 암유전체의 단일세포 분석
암은 세포 내의 유전자 변화로 인해 비정상적으로 과다하게 증식하는 질병이며, DNA의 손상, 복구, 진화 등과 관련되어 있어, 암세포 군집의 특성은 세포 수준에서 보여줄 수 있는 유전적 다양성과 불균일성의 전형적인 예라 할 수 있겠다. 따라서 단일세포 유전체학의 장점이 가장 많이 드러날 수 있는 연구 분야로 생각된다 [Van Loo 2014]. 생검으로 얻어진 샘플에는 암세포의 암 진화 과정에서 서로 다른 변이를 획득한 클론 집단들이 있으므로, 한 암조직의 여러 부분에서 채취한 샘플로 각각 시퀀싱하여 그 차이를 연구할 수 있는데, 이제 단일세포로 나누어서 그 차이를 볼 수 있게 되었다 [Wang 2014]. 또한 단일세포 시퀀싱을 이용하여 암 조직에서 떨어져 나와 혈관 혹은 골수(bone marrow)에서 발견되는 Circulating Tumor Cell (CTC), disseminated tumour cells (DTC)를 연구한 논문도 있다 [Ramskold 2012, Ni 2013].
7. Bioinformatics for single cell omics study
기본적으로는 한 개의 평균값 대신 수백 ~ 수천 개의 개별 값의 분포를 얻게 되므로 이를 대상으로 한 군집 연구 (population study)가 가능하며, 기본적으로 다변량 분석이 수행되어야 한다 [Liberali 2015]. 그러나 에러 및 노이즈에 대한 부분을 고려해야 하는데, 전장유전체 증폭 및 전장발현체 증폭 (WGA, WTA)에 사용된 효소와 프로토콜의 특성에서 오는 위음양성 (false positive, false negative)을 고려해야 한다. Microdroplet의 경우 각 cell을 index 하기 위해 사용된 UMI filtering의 처리도 필요하다. 전사체 분석에서는 단일 세포 수준이기 때문에 고려해야 할 부분들이 조금 더 생겨나는데, 이를테면 QC에서는 각 세포 내의 RNA가 degradation 되었는지 판단하고, 필요하다면 분석에서 제외해야 하며, 판단하기 위한 과정에서 spike-in control이 필요하게 된다. 또한 세포별 발현량을 어떻게 normalization 할지에 대해서도 생각해야 한다 [Stegle 2015]. 그리고 각 세포의 발현량 정보로부터 유전자 발현 네트워크를 추론하는 도구들을 새로 개발해야 할 수도 있겠다 [Benett 2015]. 단일세포로부터 genome 과 tranome, 더 나아가 epigenome 정보를 같이 얻을 수 있게 되므로 - 하나의 단일세포에서 DNA, RNA를 모두 시퀀싱하는 것도 가능하다 [Macaulay 2015] - 이 데이터들의 통합 분석에 대한 고민 역시 필요하게 된다.
8. 참고문헌
☞ 자세한 내용은 내용바로가기 또는 첨부파일을 이용하시기 바랍니다.
지식
동향