

기술동향
ASHG 2016 참관기
- 등록일2016-11-30
- 조회수6104
- 분류기술동향
-
자료발간일
2016-11-10
-
출처
한국유전체학회
- 원문링크
-
키워드
#ASHG 2016 참관기#참관기#유전학 학회
출처 : 한국유전체학회
ASHG 2016 참관기
테라젠이텍스 김태형이사
전 세계 최대 규모 유전학 학회인 “ASHG 2016 컨퍼런스”가 올해는 캐나다 밴쿠버에 있는 컨벤션센터에서 10월 18일 부터 10월 22일까지 5일간 진행되었다. 학회가 있는 일주일동안 비가 계속 내려 좀처럼 맑은 날씨를 볼수가 없었지만 경치가 좋기로 유명한 “캐나다 플레이스” 바로 옆에 학회장이 있어 멋진 바다 전경과 함께 평생 처음보는 큰 유람선이 학회장 사이로 다녔고 여러대의 수상경비행기가 바다와 하늘을 넘나들면서 이착륙하는 멋진 관경을 학회 기간 내내 선사하였다.
그리고 캐나다의 국민 커피 “TimHolton”을 마시면서 흥미진진한 세계 각국의 유전학 및 유전체 전문가의 학술발표를 보면서 공부할수 있어 이번 ASHG 학회 참석은 너무 좋았다. 학회 중간에 외국에 계신 한국인 연구자 분께서 소속된 잭슨랩 리셉션에 저희 팀을 초대해주셔서 캐나다에서 잡힌 신선한 재료로 만든 연어요리와 빙하수로 만들었다는 청량감 있는 캐나다 맥주를 마시면서 아주 신나는 경험도 하게 되었다. 그리고 한국인의 밤 행사에 참여해 많은 연구자분들과 교류하는 시간도 가질수 있어 알찬 5일을 보내고 돌아올수 있었다.

ASHG(The American Society of Human Genetics)는 1948년에 처음 학회가 설립되었다. 현재 약 8,000명의 정식 회원을 두고 있으며 이번 “ASHG 2016”는 66번째 개최되는 컨퍼런스였다.
이 학회에서는 기조, 초청, 일반 발표로 3분류로 나눠져 5일간 약 330건의 구두 학술 발표가 진행이 되었으며 약 3,100건의 포스터 학술 발표가 66개국가에서 참여한 수천명의 연구자들에 의해 진행이 되었으며 이번 학회 참석한 참여자만 약 2만명이 넘는 세계 최대 규모의 인간 유전학 컨퍼런스가 진행이 되었다.

학회 기간동안 진행된 약 3500건의 구두 발표와 포스터 발표 내용을 들여다 보면 실제 환자 데이터를 다루는 유전체 임상결과를 발표하는 비중이 약 30% 이상이였다. 실제 임상에 적용한 사례를 발표하는 학술 연구가 무척 많았으며 특히 2007년 부터 시작해 사람의 질병연구를 위해 사용하기 시작한 NGS툴을 이용한 연구가 약 10년 정도 흘러가면서 이젠 완전히 자리잡아 본격적으로 수 많은 환자들을 대상으로 그 환자들의 생명을 구하거나 치료방법을 제안하기 위한 도구로 정착되어 활용되어지고 있는 것을 실제로 보면서 학회내내 매우 흥분되었다. 한국도 올해 “NGS 임상검사실 제도” 가 실행이 되면서 몇 년안에 이런 대규모 환자 임상 검체를 활용한 의미있는 학술 발표에 한국의 연구자들도 대거 발표할 기회가 생길수 있을것 같아 매우 고무적이였다. 여튼 유전체라는 툴이 얼마나 유익한지를 과학적 방법론으로 증명하기 위해 각국의 연구자들이 엄청난 노력을 기울이고 이를 잘 정리해 연구결과로 발표하면서 학회 슬로건의 “Sharing Discoveries, Sharing Our future” 말처럼 돈 주고도 살수도 없고 어디에서 얻을수 없는 아주 값진 경험을 여기에서만 공유하는 구나 하는 느낌을 강하게 받았다.

이번 컨퍼런스의 주요 내용을 4개의 키워드로 정리하면 “대규모 데이터 공유”, “임상 적용”, “다양한 질병관련 변이”, “생물학적 기능”이 될것으로 보인다.
이번 학회에는 깜짝 놀랄만한 규모의 다수의 게놈 빅 데이터(DiscovEHR, GnomAD, FLOSSIES, TOPMed)가 공개 되었다. 이들 하나하나 자세히 들여다 보도록 하겠다.
- DiscovEHR: 대규모 1백 30만명 EHR 데이터와 25만명 WES 데이터 생산 프로젝트
DiscovEHR의 경우는 “가이징거 의료 시스템 (Geisinger Health System)” 에 모여 있는 1백 30만명의 EHR 정보를 가진 환자 중에 매년 5만명씩 약 5년간 25만명의 WES (Whole Exome Sequencing) 데이터를 확보하는 것을 목표로 시작이 되었다. 그 중에 현재 12만명에게 유전체 분석 참여 동의서를 받았으며 이번에 14년간 추적된 EHR 데이터 기반으로 >8,000 건의 “quantitive & binary trait”가 포함된 50,726 WES 데이터를 생산을 완료했다. 이 데이터들을 관련된 학술 연구 발표를 했으며 이 모든 방대한 데이터들은 “DiscovEHR Browser”를 통해 공개 되었다. (http://discovehrshare.com, allele frequency 0.1%까지 확인 가능)
1차로 생산된 이들 8,000건의 표현형 정보가 맵핑된 약 5만명의 WES 데이터들은 대부분 백인들로 구성이 되어 있어 한국인을 대상으로 활용하기에는 조금 제약은 있을듯 하다. 하지만 배울점이 많은 프로젝트임은 분명하다. 제가 가장 의미있게 본것은 약 48% 정도 되는 약 26,000명 정도는 아주 가까운 혈연 관계로 추정되는 샘플들이어서 잘 분석하게 되면 임상학적으로도 유전학적으로도 매우 중요하며 현재까지는 “Genome-Phenome”의 관련성을 더 정밀히 밝혀 낼수 있는 가장 큰 데이터셋으로는 DiscovEHR 프로젝트에서 생산된 이 빅데이터가 유일해 보였다.

WES 데이터 생산은 “리제네론 유전학 센터(Regeneron Genetics Center)”에서 맡아서 연구레벨의 데이터 생산을 진행을 했으며 이 프로젝트에서 가장 핵심 조직인 2007년도에 구축한 가이징거의 “MyCode community”를 통해 “clinical validation” 수준의 결과를 만들어 환자들에게 결과를 돌려주는 “Return-of-results program”을 이미 진행 하고 있었다. 약 5만개의 WES 데이터를 분석해보니 약 18만개의 “기능 손실 변이 (LoF variant) 가 발견되었으며 이 중에 믿을수 있는 “Curated LoF carrier”가 발견된 1,415명 중 3.5%인 49명에게서 ACMG에 정한 56개 유전자에 본인들이 20개 유전자를 더 추가해 76개 유전자(actionable genes)를 대상으로 “actionable finding”에 대해서만 “CLIA confirm”된 결과를 최종적으로 43명의 참여자들에게 전달했다고 한다.
그래서 이 시스템이 루틴하게 돌아가고 있는것인지 확인차 MyCode community 를 좀 조사해보니 계속해서 서비스를 하고 있으며 건강한 참여자들을 대상으로 처음에는 연구용으로 WES 시퀀싱한 결과를 진단 수준의 검증 프로세스를 거쳐 확인된 “actiona-ble finding” 이 있는 진단 결과를 참여자에게 전달한 건수는 현재까지 MyCode의 “Re-turn-of-results program”에 참여한 10만명 중에 총 166명이나 된다고 한다. (관련자료: http://www.geisinger.org/for-researchers/partnering-with-patients/includes/pdf/84015-6-Results_Returned-Oct2016_rev1.pdf)
이때 성인은 결과를 본인의 결정에 의해서 받아갈수가 있다고 하지만 미성년인 아이들의 경우는 진단 결과를 어떻게 전달 해야하는지에 대해서는 분명히 이슈가 있다. 그래서 실제로 가이징거팀에서는 건강한 아이들을 대상으로 유전체 진단 결과를 알려줘야 하는지에 대한 설문 조사를 실행해보았다고 한다. 전문가, 3그룹(부모 그리고 청소년)의 의견이 각각 너무 달랐다.
전문가 그룹의 의견은 유전체 진단 결과가 만약 actionable한 질병 진단 결과라면 성인에 국한해서 결과를 줘야 한다고 한 반면 아이의 부모의 경우는 actionable하든 안하든 모든 결과를 부모가 받기를 원했다. 당사자인 청소년의 경우는 성인 발병성(adult onset)질환과 관련된 진단 결과일 경우는 거의 대부분의 청소년들은 성인이 될때까지는 결과를 받지 않기를 원했다고 했다. (미성년자에 있어 결과지 전달하는 프로세스에 좀 더 주의를 기울여야 할것 같다.)
개인적으로 매우 궁금해했던 건강한 성인을 대상으로 유전체 진단 여부에 따라 의료비용 증감 효과가 어떨까인데 실제로 이번 학회에 발표가 있어 설명하면, 9명의 일차진료 의사들을 통해 무작위로 환자 100명을 선별해 유전체 시퀀싱을 했을 경우와 기존의 가족력만 가지고 진료했 을 경우 의료 비용에 있어 어떤 차이를 보이는지 알아보았다. 그 결과 검사후 유전체 시퀀싱을 가지고 진료를 한 그룹이 그렇지 않은 그룹보다 평균 67달러에서 77달러로 의료비가 15% 상승하는 것을 보았다. 그리고 이 결과를 가지고 6개월 동안 팔로업을 진행했을 경우 평균 약 456달러에서 616달러로 35% 정도로 의료비용이 급상승하는 것을 볼수가 있었다. 하지만 6개월 동안 팔로업한 13명의 질병변이 (pathogenic variant)를 가진 참여자들 중에 놀랍게도 4명은 지속적인 팔로업 진료와 함께 EHR 데이터 검토를 하게 되면서 그 질병변이아 관련된 phenotypic 증거가 발견되어 졌다고 한다. 결과적으로 본다면 건강한 성인들을 대상으로 유전체 분석을 통해 팔로업 하는 것은 의료비가 다소 증가 되는 부분은 분명히 있었지만 예방 가능한 유전성 질병이 나에게 있는지 여부를 미리 알고 대처할수 있는 추가적인 헬스케어 툴로써 그 가치가 매우 높다는 것을 알수 있었다.
학회내 DiscovEHR 팀이 아닌 다른 연구팀 발표중에 의료비용의 증감에 대한 네덜란드 연구팀에서 발표한 내용을 보게 되면 DiscovEHR팀에서 확인한 것처럼 건강인을 대상으로한 것이 아닌 정말 유전질환이 의심이 되는 환자 371명을 대상으로 WES을 수행하고 헬스케어 비용이 어떻게 증감이 되는지 조사한 결과를 보면 양성 결과의 경우는 환자당 2506달러의 의료비가 감소했으며 음성 결과의 경우는 약 2203달러 감소가 되었으며 VUS의 경우는 2428달러가 감소되었음을 볼수가 있었다고 한다. 즉 환자들을 대상으로 WES을 수행할 경우 추가적인 헬스케어 비용을 상당히 줄일수가 있으며 특히 양성 진단 결과의 경우는 WES을 통해 원인을 찾았기 때문에 추가적인 불필요한 검사를 받지 않아도 되기 때문에 좀 더 많은 의료비용 감소 효과를 보이는 것을 볼수가 있었다고 한다.
이번 컨퍼런서에서는 DiscovEHR와 연계해 다양한 기관에서 약 24건 정도의 학술 구두 & 포스터 발표가 있었으며 오바마가 발표하고 이제 막 진행중인 “정밀의학이니셔티브(PMI)”의 가장 좋은 선행된 예로써 우리도 꼭 배우고 벤치마킹해야할 가까운 미래에 진행될 대규모 정밀의학 코호트 연구의 가장 좋은 모델이라 할수 있을것 같았다.
이번 DiscovEHR 연구는 최대 규모의 유전체와 EHR 데이터가 결합이 되어 대규모 인구집단 및 가족 그리고 다양한 임상 표현형 데이터를 기반으로 어떻게 효율적으로 다양한 연구를 진행할수 있고 그 결과들은 환자들에게 문제가 되지 않고 도움이 되게 어떻게 리턴하고 다른 연구자들에게는 어떻게 개인비식별 데이터로 전환해 연구데이터를 공유하고 데이터 자체의 생산성을 높이는지에 대해 배울수 있는 거의 완벽한 프로세스를 보여주는 게놈 관련 코흐트 프로젝트였다.
- GnomAD: 사상 최대 규모의 14만명 게놈 빅 데이터 공개.

이번 컨퍼런스의 하이라이트는 브로드연구소(Broad Institute)의 맥아써(MacArthur)의 사상 최대 규모의 유전체 데이터베이스인 GnomAD(노마드) 였다.
2년전에도 맥아써 그룹은 ExAC v1.내 6만명의 WES 데이터를 아무 조건 없이 전격적으로 공개해 전 세계 연구자들은 깜짝 놀랐었다. 왜냐하면 NGS로 연구용 데이터 또는 암질환 또는 유전성질환 환자들에게서 나오는 데이터를 생산해 분석하다보면 무수히 많이 발견되어지는 불필요한 변이들 때문에 골치가 아픈데 이를 필터하는 것으로 그때 당시 가장 유용한 데이터로 1000 genome을 사용했는데 이 데이터 보다도 수백배 더 성능이 좋은 데이터베이스를 활용할수 있게 되었기 때문이였다.
이번 ASHG에서도 맥아써가 직접 나와 ExAC v1. 보다 두배나 규모가 커진 ExAC v2.와 WGS데이터가 통합된 GnomAD 14만명(126,216 WES과 15,136 WGS) 유전체 데이터를 공개하면서 맥아써가 슬라이드를 넘길때마 학회장이 술렁술렁될 정도로 참석한 연구자 대부분이 매우 흥분이 되어 있었다. 참고로 ExAC과 GnomAD는 별개의 독립적 시스템이 아니라 ExAC v2. (126,216 WES)은 GnomAD (ExAC v2. + 15,136 WGS)에 포함되어 있는 형태이다.
자세히 들여다보면 WES 60X 그리고 WGS 30X 이상되는 데이터를 GnomAD 사이트(http://gnomad.broadinstitute.org/)를 통해 공개 했으며 40Tb나 되는 VCF는 올해 말까지는 QC 작업을 마무리 짓고 다운로드 가능하게 모두 공개하겠다고 발표했다.
이 데이터베이스의 의미를 다시 한번 더 요약해서 설명하자면 휴먼의 게놈과 코딩 전 영역에 있어 우리가 알고자 하는 변이들에 있어 “allele frequency” 정보를 가장 정확하게 얻을수 있는 데이베이스를 다른 그룹이 아닌 같은 연구 그룹에서 2배 이상으로 늘려서 2년 연속 두번 공개 한것이다. 당분간은 “노마드(GnomAD)” 를 뛰어 넘을 데이터베이스는 없을듯해 보인다.
게다가 연구자들은 자유롭게 “노마드”를 이용해 맥아써 그룹에 물어보지도 허락 받지 않고 마음대로 논문을 작성하고 출판할수 있다고 위트있게 발표를해 학회장내 웃음꽃이 피었다. ^^*
그리고 중요한 부분은 노마드의 특성상 WES과 WGS 데이터를 통합함으로써 두 시퀀싱 방법에 따른 각각의 장점을 더 잘 활용할수 있게 되었다는 것이다. 특히 WGS의 상대적으로 low depth인 문제점을 WES의 “고심도 해독(high depth sequencing)” 으로 보완하고 WES의 GC 컨텐츠가 너무 낮거나 높아 엑솜 캡처가 잘되지 않는 유전자 코딩 영역이 가끔 발생하게 되는데 그런것에 치우침이 상대적으로 덜한 WGS으로 영역을 잘 커버할수 있게 되는 효과가 있어 잘 상호 보완되는 데이터가 통합되게 되어 활용도는 더 높아 질듯 하다.
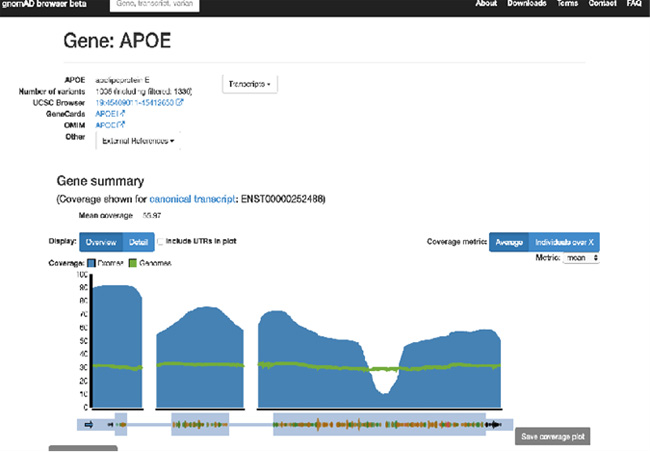
(그림은 High GC region인 APOE 유전자의 3번째 엑손이 WES의 커버리가 매우 떨어졌으나 WGS으로 보완되는 한 예를 보여주고 있다.)
하지만 인구집단이 많이 빠져 있는 상황이며, 특히 우리 한국인이 속해 있는 동양인(East Asian)은 전세계 인구의 1/3인데도 불구하고 여기 노마드 데이터에서는 1/14(1만명)정도 밖에 되지 않으며 중동인(Middle Eastern)들은 아예 빠져 있는데 여기에 대해서 맥아써의 의견은 앞으로는 더 다양한 인구 집단을 늘려가겠다고 해서 지금 버전도 엄청나지만 다음 버전의 데이터가 더 기대가 되는 상황이다. 그리고 현재는 GRCh37/hg19에 맵핑되어 있는 데이터만 공개되어 있는데 추후 40Tb VCF와 함께 최신 레퍼런스 게놈인 GRCh38 버전에 맞춰서 공개하겠다고 했다. 크리스마스 선물로 연말을 기다려 봐야겠다.

맥아써의 노마드 공개 이후 바로 뒷이어 다른 세션에서 발표한 “Anne O’Donnell-Luria”의 발표의 경우는 EaAC 게놈 빅데이터를 통해 새롭게 발견한 재미난 이야기를 풀어냈다.
우리가 임상 변이 데이터베이스라고 하는 ClinVar, HGMD 등의 변이들 그중에서도 우리가 질병변이(pathogenic variant)라고 생각하는 매우 드문 변이(rare variant) 들이 ExAC과 같은 수만명의 인구 집단 빅 게놈데이터에서 확인해 보니 이미 빈번히 발견되어지고 있어 결과적으로 pathogenic이 아닐 가능성이 매우 높다는 이야기를 했다. ClinVar와 HGMD의질병관련 변이 들의 무려 20% 정도가 ExAC 에서 1% 이상의 빈도로 발견되어 지고 있음을 알게 되었으며 즉 질병관련 변이들로 잘못 분류된 것으로 추정된다고 발표하여 기존의 그런대로 가장 신뢰하고 있는 ClinVar 및 HGMD까지도 아직 중간단계의 아주 아주 미완성의 질병변이 데이터베이스임을 확증짓는 이야기여서 크게 이슈가 되었다.
여튼 이번 ExAC를 통해 이번에 1만개 이상의 변이들을 ClinVar에 재분류되거나 추가적으로 등록되었다고 한다.
그리고 약 140개 이상의 ClinVar pathogenic variant들은 ExAC내 특정 인종에서는 매우 빈번이 발견되고 있어 그 때문에 질병변이가 아닌 것으로 다시 재분류되었다고 한다.
전체적으로 맥아써 그룹에서만 ExAC 데이터 관련해 총 9개 학술 발표가 있었고 이번 ASHG 학술 발표에서 ExAC데이터베이스를 언급하거나 활용해 연구한 발표건수가 95건 정도 있을 정도로 ExAC 자체가 이번 ASHG 학회 전반적으로 크게 부각되어 있었고 여기서도 ExAC 저기서도 ExAC하는 분위기였다. 특히 소셜네트워크(트위터, 페이스북)공간 에서는 많은 학회 참여자들이 이번 노마드 데이터베이스 공개와 관련한 찬사와 이 브라우저에 들어가 직접 이용하고 경험한 의견을 주고 받는다고 한동안 노마드 이야기만 논의되는 것을 볼수가 있었다.
이들 발표를 종합해서 보면 더 이상 우리가 알고 있는 많은 수의 potein-truncating 변이들은 ExAC에서 보게 되면 더 이상 LoF(Loss of function)가 아닌 어느 정도 질병에 intolerance 하다고 이야기 하고 있다.
- 최대규모의 유방암 관련 정상인 컨트롤 게놈 데이터베이스 FLOSSIES
워싱턴 대학교(University of Washington)의 Mary-Claire King은 “Color Genomics”라는 회사와 “Women’s Health initiative”와 함께 공동 연구 프로젝트로 “cancer free control”을 제대로 확보하기 위해 70세 이상의 평생 단 한번도 암에 걸리지 않은 여성 약 1만명(유럽계 미국인 7,325명, 아프리카계 미국인 2,559명)을 대상으로 gremlin DNA에서 BRCA1/2 등 유방암과 관련되어 있다고 판단되는 27개 유전자 패널(modified BROCA panel)을 이용해 NGS로 >250X 이상의 시퀀싱 데이터를 생산하고 분석해 “Fabulous Ladies Over Seventy (FLOSSIES)” 정상인 컨트롤 데이터베이스(https://whi.color.com/)를 만들고 공개를 했다.

이 데이터베이스의 목적은 BRCA1/2를 포함해 유방암 환자 및 의심환자에서 계속적으로 발견되는 Variant of Uncertain Significance (VUS) 를 잘 분류하기 위한 목적이 가장 큰것으로 보인다.
한국에서도 2016년 말부터 NGS 임상검사실이 실행이 되면 특히 유방암과 난소암 관련 여성 유전성 암질환에 관련해서는 질병관련 변이와 그렇지 않은 변이들을 잘 구분해서 진단을 해야하는데 이런 “cancer free control” 데이터는 유전성 암질환 스크리닝 서비스 함에 아주 크게 도움이될것으로 보인다.
- 세계에서 가장 큰 규모의 WGS 데이터베이스 TOPMed 공개
최근 크레이그 벤터(craig venter)가 1만명 WGS 데이터를 발표해 큰 이슈가 되었는데 몇 달 사이에 그 발표가 무색해질만큼 이번 학회에서는 그것보다 5배가 더 큰 52,411명의 휴먼 WGS 데이터를 TOPMed (Trans-Omits for Precision Medicine) 라는 이름으로 전격적으로 공개해 깜짝 놀라게 했다.
미시간 대학과 워싱턴 대학 두 기관에 데이터 코디네이션 센터 역할을 맡아 24개 다양한 연구PI들이 공동으로 TOPMed 프로젝트에 참여해 WGS으로는 맥아써 그룹의 GnomAD 보다도 훨씬 높은 퀄리티의 WGS 데이터를 생산하고 분석 및 관리 하고 있었으며 2주전 학회 발표 당시만 해도 52,411명의 데이터가 생산되었는데 이 글을 쓰고 있는 지금 현재 2016년 11월 4일 기준으로 55,179명의 데이터가 생산된 것처럼 매일 매일 다르게 급격하게 데이터가 늘어나고 있으며 실시간으로 데이터가 생산되어지는 내용을 아주 투명하게 공개하고 있는 것을 볼수가 있었다. (http://nhlbi.sph.umich.edu/report/) 여기 데이터의 인종 그룹을 보면 유럽계 미국인: 54%, 아프리카계 미국인: 26%, 남미/라틴계: 10%, 아시아: 7%, 나머지: 3% 비율로 구성 되어 있다.
생산되는 시퀀싱 데이터의 퀄리티는 최상으로 유지되며 평균 약 38X로 99% 게놈 커버리지와 0.34% 이하로 DNA 오염을 유지하며 관리를 한다. DNA 염기수로 계산해보면 약 7,000조개의 DNA를 생산했다고 볼수가 있다. 우리가 WES 및 WGS을 시퀀싱하고 나면 변이 분석 단계에서 항상 사용하는 컨트롤 데이터베이스인 1000 genome 보다 도 규모가 최소 100배 정도 더 커다고 보면 될듯 하다.
현재 분석까지 완료된 WGS 데이터는 18,877 건이 있으며 이를 통해 유래된 VCF 파일로는 약 20TB 정도 된다고 한다.
하지만 실제로 공개된 데이터는 dbGaP에 엠바고 없이 약 8,000명 이상의 데이터가 학회가 있는 주에 공개가 되었으며 Bravo browser(http://bravo.sph.umich.edu/)를 통해 약 14,559명의 WGS 데이터에서 유래된 변이 정보를 브라우징 할수가 있게 되어 있다.
데이터 공개 방식이 좀 다양하고 복잡하긴 하지만 공동으로 진행하는 24개 연구 주체들과 문제 없이 커뮤니케이션 하고 협력하면서도 빠르게 데이터를 공개하는 방식으로 현재까지는 가장 성공적인 공동 유전체 프로젝트로 보인다.
발표가 끝나고 질문중에 구조변이 (SVs) 정보는 왜 공개하지 않느냐는 질문에는 답변으로는 지금 현재는 SNPs/Indel만 공개하고 있고 구조변이는 데이터의 재현성 때문에 아직 핸드링 하지 않고 있지만 추후 어떻게든 분석 기술을 발전시켜 우선순위로 분석을 하고 공개하겠다고 했다.
여기 TOPMed 프로젝트에 참여한 기관을 보면 미국의 왠만한 쟁쟁한 유전체 그룹들은 모두 참여하고 있으며 특히 베일러, 브로드, 미시간대학, 워싱턴대학 등이 주도적으로 그리고 경쟁적으로 WGS 데이터 생산 및 공유에 참여하고 있음을 느낄수가 있었다.
- 중국 BGI의 1.5M Chinese Millionome Project
중국 BGI의 NIPT 서비스인 NIFTY를 이용해 서비스된 약 1.5백만건의 low-pass WGS(0.06X-0.1X) 임상 데이터를 이용해 연구를 진행중에 있다. 이번 학회에서는 약 14만명의 데이터를 분석한 결과 발표가 있었다.
이 프로젝트의 경우는 기존 다른 대규모 코흐트 연구(1000Genome, Genome England, ExAC, DiscovEHR, TOPMed, HLI, 등)에서 제한적인 아시아 인구 집단을 가지고 연구를 했다면 이 “Chinese Millionome Project“를 통해 연구되어지는 1.5백만명은 거의 대부분이 중국 내부(1.4백만명) 에서 일부는 아시아 다른국가(10만명)에서 서비스를 의뢰해 진행이 되는 건이라 한국인과 가까운 인종그룹의 대규모 데이터가 있어 우리 입장에서는 매우 가치가 있는 데이터로 여겨진다.
그렇지만 이번에 ASHG에서 발표한 다른 여타 대규모 코흐트 프로젝트는 모두 공개한 반면에 데이터를 아직은 공개하지 않고 컨설시움내에서만 공유하고 있어 데이터 접근성이 전혀 없다는 문제가 있다. 현재까지 NIPT는 전 세계 5백만명 정도가 서비스를 받았고 그 중에 3백만명 정도는 중국에서 서비스가 진행이 되었다고 한다. 또 그 중에서 50%인 1.5백만명을 BGI NIFTY를 통해서 서비스가 되었고 이 모든 데이터들은 Chinese Millionome Consortium을 통해 다양한 연구가 진행이 되고 있다고 했다.
이 연구 내용을 요약하면 본인들은 150bp~200bp 되는 DNA 조각들을 약 35bp~49bp 리드 길이로 5-8백만 리드 정도 읽어 들여 약 0.06~0.1X 정도의 low-pass WGS을 통해 산모 혈액에서 유래한 태아의 5-15%되는 DNA를 산모 DNA 유래 DNA와 같이 생명정보학적 데이터 분석을 통해 선천적 염색체 이상을 진단하고 있으며 이때 이러한 빅 데이터들이 쌓이게 되어 연구에 활용되고 있다고 한다. (우리나라는 생명윤리법상 익명성이 보자된 유전체 검사 결과임에도 불구하고 모두 폐기 하는 것으로 명문화 되어 있어 이러한 빅데이터가 쌓일수 있는 여건이 안되어 있어 제도적인 개선이 필요해 보인다.)
이 방대한 데이터들은 BGI가 보유하고 있는 엄청난 서버 컴퓨터들을 하둡 클러스터로 엮어 데이터 분석에 이용했으며 14만명의 데이터를 핸드링 함에 있어서 약 1PB 정도 되는 공간이 필요했다고 한다. 워낙 낮은 뎁스로 읽어 들인것이라 이 데이터 특성을 보면 보통 30X로 읽어서 처리한 패턴과 2 read 이상이 맵핑되는 영역이 거의 없어 1 read가 붙는 영역이 약 93% 정도가 되며 이러한 데이터를 가지고 연구를 진행했으며 BRCA variant들을 대거 찾았으며 중국과 유럽인에서 발견되어지는 변이 빈도가 크게 차이가 나는 것을 알수가 있었다고 한다.
특히 BRCA2에서 발견되어지는 변이들의 경우는 유럽인들보다 중국인들에서 더 빈번히 발견되어지는 것을 보고 중국에서 유럽으로 유방암 관련 인구집단이 이동했다고 추정한다고 슬쩍 말하고 있었다.(뉘양스가 인류의 기원을 중국이라고 말하고 싶은것 같아 보였다.) BRCA 이외에도 Hearing loss와 Beta thal 관련 잘 알려진 질병관련 변이들도 다수 스크리닝 되었으며 유전성 암과 관련된 “incidental findings”도 다수 스크리닝되었다고 한다.
이러한 중국내 인구 집단 유전체 연구를 통해 지역별 그리고 다양한 인종 그룹에 대해서 유전적 차이와 관계성을 더 명확하게 알게 되었으며 이렇게 0.1X로 생산된 데이터지만 14만명의 유전체를 합쳐서 분석해보니 imputation할때는 정확도가 약 0.6 정도로 나쁘지 않게 나왔다고 한다. 그리고 이 “Chinese Millionome Consortium”은 전 세계적으로 열려있고 참여할수 있다고 발표자 “Xin Jin”이 말했다. 하지만 현재까지 BGI는 유전체 데이터를 단 한건도 공개하지 않았다.
- ClinVar 분석 및 데이터베이스 업데이트
코딩 유전자내 미스센스 돌연변이가 전혀 발견되지 않은 유전자를 CCRs(Coding Constrained Regions)라고 정의하고 ClinVar 데이터베이스에서 확인을 해보니 질병관련 변이들의 50%가 이 CCRs 관련 유전자에서 발견되었으며 “필수 유전자(Essential Gene)”라고 잘 알려진 유전자들은 대부분 상위 1~5% CCRs에 들어가는 것이 확인되어졌다. 특히 심각한 지적 장애(Severe intellectual disability)관련 해서 발견되어지는 de novo 변이 같은 경우는 CCRs에서 더 많이 발견되어 졌다고 한다. 결과적으로 해석해보면 상위 1~5% 정도의 CCRs에 변이가 발견될 경우는 임상적으로 예후가 좋지 않을 가능성이 매우 높고 아주 초기 영유아 단계에서 발병되는 질병과 관련된 변이가 발견되어지는 경우가 많으며 이들 유전자의 기능을 검증해야 하며 이들 유전자들의 기능들을 알아가는 것이 매우 중요한 연구가 될것이라고 “Jim havrilla”가 발표를 했다.

ClinGen 부스에서는 큰 전시 패널을 만들어 ClinVar 데이터베이스에 질병학적으로 해석 가능한 변이들을 아무 조건없이 기부(등록)한 주요 임상서비스 센터들을 모두 공개했다. GeneDx와 Ambry Genetics, Invitae, Partners healthcare 등이 대표적인 임상 유전체 진단 서비스 회사 및 기관들이며 각자 수만개의 임상적으로 의미있는 변이들을 질병을 연구하는 전 세계 연구자 및 환자진단을 돕기 위해 공개하였다. (아직도 최대 유전자 검사를 수행하는 미리아드는 ClinVar에 단 한건의 변이도 등록하지 않고 있다.)
학회가 진행되는 중에 일루미나까지도 현재까지 임상유전체서비스(clinical genome service, Understand Your Geome) 를 통해 완료된 참여자 >1,500명 대상으로 발견된 >9만 5천개 변이 들을 ClinVar에 추가로 등록 하기로 결정했다는 발표가 있었다.
이렇게 각 연구 기관 및 진단 회사들이 자신들이 특정 인구집단을 연구하면서 그리고 환자 진단 서비스를 하면서 발견되어지는 변이들을 기꺼이 공개하는 것을 보면 조만간에 애매하게 분류되어지는 변이들이나 VUSs 들이 대부분 사라지지 않을까 한다.
- 세계 최대 규모의 유전체 연구를 진행하는 “23andme research”

“23andme research”는 23andme가 DTC로 서비스한 150만명 유저들을 대상으로 유전자와 질병 관계성을 밝히는 연구에 참여할지 여부를 물으니 놀랍게도 약 80%의 유저들이 참여 하겠다고 결정을 했다고 한다. 그리고 이들은 기꺼이 300개나 넘는 설문지 문항에 응답했다고 한다. 이렇게 확보된 “유전체 + 설문 결과”를 바탕으로 연구자들에게 데이터를 오픈하고 각종 질병 및 표현형과 관련된 GWAS 연구결과를 도출하는 방식으로 수십개 연구가 진행되고 있고 이번 컨퍼런스 동안만 “23andme research”를 언급하거나 이용해 발표한 학술 발표만 약 20건 정도 되는 것을 볼수가 있었다.
23andme의 “Richard Scheller”가 학회에서 발표한 내용에 따르면 백만명이 넘는 23andme 유저중에 동양인은 단 4% 정도 밖에 되지 않는다고 하는 안타까운 이야기를 들었다. 23andme 데이터를 가지고 동양인을 대상으로 뭔가를 연구 하는 것은 상대적으로 제한적일수도 있어 보인다. (연구소 뿐만 아니라 회사까지도 동양인의 유전체 데이터가 적은 것을 보면 서양인들에 비해 동양인들이 유전체 연구 및 개인 유전체 서비스 참여에 소극적인 것으로 보인다. 참고로 저는 이미 10년 전부터 참여했습니다.)
- 900만명 환자 EHR 데이터를 기반으로 한 유전성이 강한 표현형 발굴 프로젝트
학회 첫 기조 학술 발표로 콜롬비아 대학의 “Nicholas Tatonetti”가 약 9백만명의 환자 EHR 데이터를 이용해 유전성이 강한 질환과 표현형을 예측하는 내용을 발표를 했다. (최대 규모의 데이터라 기조 발표중에서도 가장 먼저 발표를 하게 했나 봅니다.)
이 연구가 재미있는것은 단 한건도 DNA분석을 하지 않았다는 것입니다. 단지, EHR 정보만 가지고 수천가지 표현형중에 유전성이 강한 포현형을 찾아내는 그것도 아주 정확하게 정의를 한 것에 놀라울 뿐이였다.
방법과 결론은 너무 심플했다. 익명화된 EHR내 정보중에 비상연락망 정보를 통해 가족 관계를 추정해 이를 기반으로 >500개 표현형 중에 어떤것이 가족들 내 세대를 거쳐 유전되며 그 유전성이 얼마나 강한지를 분석을 수행해 최종적으로 유전적으로 강한 49개의 표현형 정보를 결정했고 이 분석을 통해 사람의 키 같은 경우는 유전성이 77%나 있다는 결론을 내렸다.
- BioVU 프로젝트
BioVU 프로젝트를 통해 2백 5만명을 대상으로 현재는 약 22만명의 DNA를 확보했으며 약 2만 5천명의 GWAS array 데이터를 확보했으며 2017년 말까지 약 12만명의 array 데이터를 확보할 예정이 있다고 하며 궁극적으로는 유전자 기반의 phenome-wide 연관성 카탈로고를 구축하는 것을 목표로 진행되고 있다고 한다. “Nancy Cox” 발표자는 연구를 해보니 멘델리안 질환 환자의 위험도를 증가시키는 핵심은 LoF 변이 보다는 결국은 특정 유전자의 발현이 감소되는 것으로 인해 질환 위험성이 높아진다고 했다.
- 대규모 유전성 암유전자 패널 스크리닝
“G.E Tiller” 발표자는 20개의 유전성 암유전자(BRCA1/2, MUTYH, CHEK2, ATM, PALB2, PMS2, MSH6 등) 패널 을 만들어 암의 가족력이 있는 약 3천 5백명의 환자를 대상으로 스크리닝 해보았다.
그랬더니 이들 환자들이 분명히 암질환 가족력이 있음에도 불구하고 놀랍게도 절반이상인 약 58% 정도는 pathogenic 또는 likely pathogenic이 전혀 발견되지 않았으며 단지 12%만 알려진 질병 관련 변이가 발견되었다고 한다. 나머지 약 30% 정도는 관련 유전자에서 VUS들만 다수 발견되어졌다고 한다. 특히 소수인종 그룹에서는 VUS 비율이 상대적으로 매우 높게 나왔다고 한다.
즉 아직도 우리가 밝혀낸 알려진 질병 관련 변이들이 그렇게 많지가 않다는 것을 말하고 있고 아직 더 많은 연구가 필요하며 특히 더 많은 다양한 인종에서 연구가 되어질 필요가 있다는 것을 역설적으로 보여주는 프로젝트여서 의미가 있었다.
- 유전질환을 앓는 아이의 부모가 생각하는 Clinical WES
캐나다 브리티시 컬럼비아 대학의 한 연구팀은 어린 자녀를 대상으로 clinical WES을 할지 안할지를 결정할때 어떤 이슈들이 있는지를 원인 불명의 early-onset 간질 아이가 있는 가족들을 대상으로 clinical WES 서비스를 어떤 이유에서 결정을 하게 되었는지 조사해 보았다고 한다.
109 가족중 설문에 참여한 사람의 대부분은 여성이였으며 약 84%였고 79%는 대졸의 고학력자였다고 한다. Clinical WES 서비스를 받겠다는 사람들의 참여 결정 요인(중복 체크)을 보면 참여자의 92%가 내 자식의 유전정보를 알고 싶다고 했고 85%가 그 정보를 알아서 질병을 관리하기 위해서 라고 햇으며 약 72%는 과학의 발전을 위해서라고 했다. 그리고 31%는 너무 슬퍼서? 약 24%의 부모들은 아이들한테 죄책감이 느껴져서 “Clinical WES” 서비스를 수행하기로 결정 했다.
놀라운 것은 많은 희귀질환 아이를 둔 부모들 대부분이 남을 돕기 위한 애타주의적인 생각을 강하게 가지고 있어 본인들의 Clinical WES을 활용해 다른 사람들에게 도움이 되기를 바라고 있고 이런 유전학 연구에 적극 참여하고 데이터를 공유하는 것에도 적극적인것을 볼수가 있었다.
- 제네틱 카운셀링의 툴의 새로운 변화
제네틱 카운셀링 세션이 있어 참석해 발표를 들었다. 국내에서는 아직 유전체 검사가 실행되지 않고 있어 제네틱 카운셀링이 실제로 필드에서 활발하게 진행되고 있지는 않지만 미국 및 선진국에서는 이미 제네틱 카운셀링이 활성화 되어 있었다.
이미 기존의 1대1 면담으로는 유전체 기반의 제네틱 카운셀링을 커버하기에는 너무 어렵고 이런 현상에 잘 대응하기 위해서 벌써 앞서가는 연구자들을 중심으로 e-카운셀링 같은 새로운 방법이 개발되어 지고 있었다.
이미 1대1 면담 방법의 일부 기능을 대체하고 이러한 방법에 대한 효용성 평가를 하고 있었으며 이를 실제로 의료현장에서 효과적으로 적용하기 위한 언어문제, 낮은 독해력, 수리력, 문화/사회/경제적 차이와 학습 장애 차이를 완화하기 위한 e-카운셀링 시스템으로 한 차원 더 도약하기 위한 노력을 기울이고 있음을 볼수가 있었다. 우리나라에서 본격적으로 NGS 임상검사 서비스가 실행이 된다면 병목현상이 발생할 분야가 이 유전상담이 될것으로 보이는데 e-카운셀링이 대안이 될듯 하다.
- 마운트 시나이 병원의 보인자 스크리닝 테스트
마운트 시나이 병원의 확장된 보인자 스크리닝 테스트(277개 유전자 패널, 평균 500X)를 이용해 >76,000명 대상(약 83% 정도가 여성이였으며 약 7,000 커플이 테스트를 받았으며 약 38%가 백인이였으며 약 27%는 유대인)으로 ACMG 가이드라인에 따라 스크리닝을 수행해 pathogenic variant(VUS는 제외)만을 레포트하는 방식으로 서비스를 했다.
특정 질환에 가족력이 있을 경우에 한해서만 VUS를 레포팅하는 방식으로 진행이 되어졌다. 특정 몇가지 유전자에 대해서는 NGS 유전자 패널로 커버가 되지 않아 MLPA와 FragileX 그리고 제노타이핑을 수행했으며 전체적으로 보인자 커플이 88쌍이 발견되었으며 약 1/80 빈도로 발견되는 패턴을 보였다고 한다. 꽤 빈번이 보인자가 발견되고 있음을 볼수가 있으며 젊은 남녀 커플이 미리 이 정보들을 알고 가족 계획을 한다면 크게 유용할것으로 보인다.
- ASHG 한국 연구자 참여
한국인 연구자들이 참여한 학술발표(포스트)는 총 약 250여건 정도 되는것으로 보이며 국내 소속 연구자들이 약 50여건이 되며 해외 소속 한국인 연구자들이 약 200여건이 넘는 것을 볼수가 있었다. 외국에 거주하는 한국인 연구자들의 ASHG 참여도가 훨씬 더 높음을 간접적으로 확인할수가 있었다.
다음 ASHG는 2017년 11월 17일~21일까지 미국 플로리다의 올란도에서 개최될 예정입니다. 내년에 ASHG 2017에서 뵙겠습니다.
☞ 자세한 내용은 내용바로가기를 이용하시기 바랍니다.
지식