

기술동향
RAD-Seq을 이용한 생태 및 진화 유전체 연구
- 등록일2017-10-19
- 조회수6446
- 분류기술동향
-
자료발간일
2017-09-26
-
출처
생물학연구정보센터(BRIC)
- 원문링크
-
키워드
#RAD-Seq
- 첨부파일
 pdf_0002828.pdf
pdf_0002828.pdf
출처 : 생물학연구정보센터(BRIC)
RAD-Seq을 이용한 생태 및 진화 유전체 연구
본 자료는 Harnessing the power of RADseq for ecological and evolutionary genomics. Nature Reviews Genetics, 17 (2), pp. 81-92.의 논문을 한글로 번역, 요약한 자료입니다.
[목 차]
1. 서론
2. RAD-Seq 계열의 분석
3. 오류 및 편향의 원인
4. RAD-Seq 연구 설계 방법
5. 결론
[요약문]
제한효소 관련 DNA 시퀀싱(RAD-Seq)을 기반으로 한 대용량 처리분석 방법들은 생태, 진화 및 보전 유전학에 혁명을 일으키고 있으며, 비모델 생물체를 포함한 다양한 종에 대한 수천 가지 유전자 마커의 저비용 식별 및 유전자 타이핑을 가능하게 한다. 이러한 방법들 간의 기술적 차이는 해결될 수 있는 특정 과학적 질문과 라이브러리 준비 및 시퀀싱의 비용, 결과 데이터에 내재한 편향 및 유형에 대한 유전체 연구의 모든 단계에서 중요한 고려 사항이 된다. 본 리뷰에서는 연구자들이 잘못된 과학적 결론에 도달하지 않고, RAD-Seq 데이터의 올바른 분석 방법을 선택할 수 있도록 RAD-Seq 분석 방법에 대한 포괄적인 정보를 제공할 것이다.
1. 서론
제한효소 관련 DNA 서열 분석(RAD-Seq)의 개발은 지난 10년 동안 가장 중요한 과학적 발견 중 하나로 간주되었다. RAD-Seq은 게놈을 통해 수백 또는 수천 개의 다형성 유전자 마커를 밝히기 위해 차세대 시퀀싱의 막대한 처리량을 활용하여 생태, 진화 및 보전 유전체학에 대한 연구에 활력을 불어 넣었다. RADseq은 유전체의 일부분만을 대상으로 하므로 한정된 예산에도 많은 샘플 수를 확보할 수 있으며 전장 유전체 분석에 비해 타겟하는 부분에 대한 더 높은 시퀀싱 양(depth-of-coverage)을 확보할 수 있다(이는 시퀀싱의 신뢰도를 높여주게 된다). 또한, RAD-Seq은 분석하는 생물종에 대한 참조유전체 서열정보를 요구하지 않는다는 장점이 있다. 결과적으로, RAD-Seq은 비모델 생물체에 대한 생태 및 진화 연구에서 고처리량(high-throughput) 단일 뉴클레오타이드 다형성(SNP) 발견 및 유전자 타이핑에 가장 널리 사용되는 게놈 접근법이 되었다.
RAD-Seq라는 용어는 원래 하나의 특정 방법을 설명하기 위해 사용되었지만 이후에는 시퀀싱될 유전자좌 세트를 결정하기 위해 제한효소에 의존하는 관련 기술을 모두 아우르는 것으로 확장되었다. 또한 이 방법은 때로는 ‘시퀀싱을 통한 유전자 타이핑(genotyping by sequencing)’ (GBS) 기술이라는 용어로도 분류된다. RAD-Seq은 차세대 시퀀싱을 사용하여 많은 수의 제한효소 절단 사이트에 인접한 시퀀스 데이터를 생성한다. RAD-Seq 유전자좌는 유전체 내의 모든 영역(단백질 코딩 영역과 비코딩 영역 모두)에서 발생할 수 있으며 밀접한 관련 종은 일반적으로 절단 부위의 보존으로 인해 대부분의 유전자좌를 공유한다.
지난 수년간 다양한 종류의 RAD-Seq 방법론이 개발되었으며 연구에 맞춰 다양한 유연성을 갖도록 발전되었다. 또한 다양한 생태 및 진화 유전체학 연구에서 비용과 노력을 줄일 수 있는 혁신적인 기술로 발전하고 있다. 그러나 다양한 방법이 발전해 온 만큼 잘못된 RAD-Seq 방법론의 선택은 연구 설계 및 실행에서 결과 데이터 출력에 이르기까지 게놈 연구의 모든 단계에 중대한 영향을 미칠 수 있다.
이 리뷰 논문에서는 주로 자연 개체군의 생태 및 진화 유전학에 RAD-Seq을 적용하는 것에 중점을 두지만 농업 종의 형질지도 작성과 같은 다른 RAD-Seq의 응용에도 초점을 맞출 것이다. 그리고 개발된 다양한 RADseq 기술에 대한 개요와 이러한 방법을 적용할 수 있는 다양한 연구 질문을 기술할 것이다. 또한 다양한 방법론 간의 기술적 차이가 야기하는 실험 설계 및 분석의 차이와, RAD-Seq 연구를 설계하기 위한 일반적인 고려 사항 등도 기술하고자 한다.
2. RAD-Seq 계열의 분석
RAD-Seq 기술은 몇 가지 기본 단계를 공유한다(그림 1). 모든 방법은 고분자량의 유전체 DNA로 시작하여 하나 이상의 제한효소로 절단을 시작한다. 모든 방법은 차세대 시퀀싱 플랫폼에 필요한 특정 시퀀싱 어댑터 또는 이중 가닥 올리고 뉴클레오티드를 추가(adding)한다. 사용된 효소들에 따라 차세대 시퀀싱에 최적화된 크기의 DNA 단편 길이가 조절될 수 있다.
RAD-Seq 분석방법들은 효소의 절단, 어댑터의 라이게이션, 크기 결정, 시퀀스 데이터 타입 등 여러 가지 요인들에 의해서 달라질 수 있다. 우리는 각 단계에서의 방법들 사이의 중요한 차이점과, 이러한 차이가 야기하는 ‘라이브러리 준비’, ‘결과 데이터’ 및 ‘후속 생물 정보 분석’에 대한 결과 중 일부를 논의해보고자 한다.

그림 1. 5가지 RADseq 라이브러리 준비 프로토콜의 단계별 그림. 모든 프로토콜은 하나 이상의 제한효소로 고 분자량 게놈 DNA를 분해함으로써 시작된다. 대부분의 프로토콜의 경우 시퀀싱 어댑터(oligonucleotides)는 1) 프로토콜의 초기 단계에서 연결(ligation) 단계 중 그리고 2) 최종 PCR 단계 동안 통합된 두 번째 올리고 뉴클레오티드로 추가된다. 두 번째 단계의 올리고 뉴클레오타이드 세트는 완전한 일루미나 어댑터 서열을 생성하기 위해 전체 단편의 길이를 연장시킨다. 반대로 Original RAD-Seq은 어댑터를 3단계로 추가한다. Illumina 시퀀싱의 경우, 각 DNA 조각의 양쪽 끝에 있는 어댑터가 달라야하므로 일부 프로토콜(예: 원본 RADseq, 이중 절단 RAD (ddRAD) 및 ezRAD)은 Y- 어댑터를 사용하여 서로 다른 양쪽 끝의 어댑터가 PCR 증폭된다. 다른 프로토콜(예: GBS)은 올바른 어댑터가 없는 단편이 시퀀싱되지 않는다는 원리를 이용한다. 시퀀싱에 이상적인 길이의 단편을 생성하기 위해 대부분의 방법은 일반적인 절단기(common-cutter) 효소(예: 4-6 bp 절단기)를 사용하여 광범위한 단편 크기를 생성한 후 직접 크기 선택(젤 커팅 또는 마그네틱 비드, ezRAD 및 ddRAD) 또는 간접 크기 선택(예: GBS와 같은 PCR 증폭 또는 시퀀싱 효율의 결과)을 수행한다(그림 원본 URL: http://www.nature.com/nrg/journal/v17/n2/full/nrg.2015.28.html).
? 시작 DNA
RAD-Seq 기술은 고분자량 유전체 DNA로 구성된 출발 물질(starting material)을 기반으로 최적화되었으므로 이러한 기술은 과도하게 분해된 유전체 DNA로는 제대로 수행되기가 어렵다. 예를 들어, 효소 특이적 어댑터가 없는 방법(예: ezRAD 및 CRoPS)에서는 절단 부위에 인접하지 않은 작은 DNA 조각이 시퀀싱 라이브러리에서 끝나기 때문에 불필요한 시퀀싱(비-RAD 유전자좌)이 발생한다. 일반적으로 더 많은 양의 DNA는 필요한 PCR 사이클의 수를 줄일 수 있으며 PCR 복제시 발생할 수 있는 문제를 최소화할 수 있기 때문에 대체로 권장되어 왔다. 그러나 대부분의 RAD-Seq 방법은 샘플당 필요한 총 DNA 양이 50-100 ng으로 적은 양의 DNA로도 분석이 가능하다. 한 가지 예외는 ezRAD에서와 같이 대량의 시작 DNA(예: 1-2μg의 DNA)를 필요로 하는 PCR 프리 라이브러리 준비 방법을 사용할 때이다.
? 제한효소 절단
RAD-Seq 프로토콜은 사용된 제한효소의 수와 이들 효소가 게놈을 절단하는 빈도에 따라 다르며, 서열화된 유전자좌 세트의 게놈에서의 분포와 어떻게 관련이 있는지에 따라 크게 두 가지 그룹으로 나뉜다. RAD-Seq 프로토콜과 2bRAD는 제한효소의 모든 절단 부위에서 서열 데이터를 생성하는 것을 목표로 한다. 이와는 대조적으로 다른 나머지 기술들은 특정 게놈 거리에 의해 분리된 두 개의 효소 절단 부위에 의해 생성되는 게놈 단편의 서열 결정에 의존한다. 이러한 절단 부위는 하나 또는 두 개의 효소를 사용하는지 여부에 따라 동일하거나 상이한 효소로부터 유래된 것일 수 있다. 각 방법에 대해 공통 절단기 또는 희귀 절단기 효소를 사용하여 생성된 유전자 좌의 수를 조정할 수 있다(역자: 게놈상에서 상대적으로 많은 부분을 절단하는 것을 ‘공통 절단기’, 상대적으로 희귀한 부위만을 절단하는 것을 ‘희귀 절단기’ 라고 부름).
? 어댑터 연결
RAD-Seq 기술은 어댑터가 어떻게 구성되고 DNA 단편에 연결되는지, 그리고 목표 게놈 DNA 단편(즉, 제한 효소 절단 부위에 인접한 단편)만 서열화되도록 하기 위해 어떻게 설계되는지에 따라 달라질 수 있다. 경우에 따라 어댑터는 소화 후 제한효소 절단 부위에 남아있는 특이적 단일 가닥 점착성(sticky) 말단에서만 결합(ligation)되도록 설계된다. Illumina 시퀀싱 기반의 많은 RAD-Seq 프로토콜은 시퀀싱에 필요한 어댑터 조합을 가진 단편만 PCR 증폭할 수 있도록 구조화된 Y- 어댑터를 사용할 수 있다(그림 1). 일부 기술은 라이브러리 구축을 위한 시약 비용뿐만 아니라 신뢰성을 증가시킬 수 있는 어댑터 연결 장치(예: ezRAD, CRoPS 및 RRL)용 특정 라이브러리 준비 키트를 사용한다. 이러한 특정 어댑터는 점착성 말단에 연결되지 않아 제한효소 절단 부위에 인접하지 않은 분해된 DNA 단편으로부터 서열 데이터가 생성될 수 있으므로 결합 특이성(ligation specificity)이 낮아질 수 있다.
? 크기 선택
대부분의 프로토콜의 경우, 제한 효소는 DNA를 광범위한 단편 길이의 조각들로 감소시킨다. 크기 선택 단계는 시퀀싱을 위한 이상적인 길이의 단편을 분리하는데 사용되며, 이 단계가 RAD-Seq 프로토콜 간의 주요 차이점을 이끌게 된다. 각각의 잠재적 궤적이 절단 부위 사이의 거리에 의해 결정되는 특징적인 단편 크기를 가지므로, 두 개의 절단 부위가 측면에 있는 DNA 조각을 서열화하는 모든 방법에 대해, 유전자형화될(genotyped) 유전자좌의 세트는 이 크기 선택에 의해 더욱 감소된다. 이러한 기술에서 크기 선택은 PCR 증폭 또는 시퀀싱 효율(예: GBS 및 CRoPS)의 결과로 간접적으로 또는 수동 또는 자동 젤 커팅 기술 또는 마그네틱 비드를 사용하여 직접 수행된다. 이러한 방법의 경우 라이브러리 전반에 걸친 크기 선택의 일관성은 샘플을 통해 유사한 유전자좌 세트에서 데이터를 생성하는데 매우 중요하다. 크기 선택의 불일치는 서로 다른 라이브러리에 나타나는 유전자좌의 다른 세트로 이어질 위험이 있으며 결과적으로 불필요한 시퀀싱 작업이 수행되거나 정확도가 떨어지게 된다.
대조적으로, 원래의 RAD-Seq 프로토콜과 2bRAD는 서열화될 유전자좌를 줄이기 위해 크기 선택을 사용하지 않으며, 제한효소 절단 사이트에 인접한 모든 유전자좌를 표적화한다. Original RAD-Seq 방법은 Illumina sequencing에 적합한 단편을 생산하기 위해 기계적 절단(mechanical shearing) 단계가 있는 단일 효소로 절단한 후 사용되었다. 이 접근법은 각각의 서열된 단편이 한쪽 끝에서 절단 부위를 갖고 다른 쪽에서 무작위로 절단된 단편을 가지며 각 단편에서 단편 크기의 범위가 생성됨을 의미한다. 결과적으로 크기 선택 단계는 유전자좌 세트를 더 이상 줄이지 않고 Illumina 시퀀싱 효율을 최적화하고 어댑터 이합체를 제거하는 데에만 사용된다. 2bRAD 방법은 RAD-Seq 프로토콜 중에서 유일하게 iib 제한효소를 사용하여 모든 유전자좌에서 동일한 크기의 짧은 단편을 생산한다.
? 바코딩(Barcoding)
어댑터에 내장된 바코드를 사용하면 일부 프로토콜의 라이브러리 준비 초기에 개별 샘플을 멀티플렉싱 할 수 있다. 이 멀티플렉싱은 때로는 풀링이라고도 하지만 샘플 각각을 하나의 바코드로 통합하는 풀링과 다른 개념임에 유의해야 한다. 라이브러리를 준비하는 동안 바코드된 어댑터를 각 샘플에 연결하는 즉시 샘플을 멀티플렉싱 할 수 있으므로 많은 수의 샘플을 사용하는 연구에서 후속 단계의 시간과 비용을 크게 줄일 수 있다. 라이브러리 준비 초기에 샘플을 멀티플렉싱하려면 인라인(in-line) 바코드를 사용해야 한다. 전용 키트의 어댑터에는 인라인 바코드가 없으므로 인라인 바코드에는 맞춤형 어댑터가 필요합니다. 또한 하나의 인라인 바코드와 DNA 단편의 반대쪽 끝 부분에 PCR 단계에서 추가한 Illumina 색인을 조합 바코드(combinatorial barcoding)로 사용하여 많은 기술을 사용할 수도 있다. 이 밖에도 또 다른 조합 바코딩 전략은 DNA 단편의 각 면에 하나씩 2개의 Illumina 색인을 사용하는 것이다. 하지만 이 방법은 library 준비 때 시료를 멀티플렉싱 하는 것을 허용하지 못하는 단점이 있다.
? 서열(sequence) 데이터의 유형
대부분의 RAD-Seq 기술은 현재 Illumina 시퀀싱을 사용한다. Illumina 기기는 일련의 리드 길이(현재 50-300 bp, 향후에는 더 증가할 가능성이 있음)를 제공하며 DNA 단편당 하나의 리드(read)를 생성하는 단일 종단 시퀀싱 또는 한 쌍의 ‘순방향(forward)’ 리드와 하나의 '역방향 (reverse)' 리드를 생성하는 쌍을 이루는 시퀀싱이다. 이 옵션은 모든 RAD-Seq 라이브러리에 적용될 수 있지만, 매우 짧은 리드(33-36 bp)를 생성하는 2bRAD에는 쌍을 이룬 서열 분석이 유용하지 않다. 다른 모든 방법의 경우, 순방향 리드는 제한효소 절단 사이트로부터 시작하며, 보다 긴 리드는 통상보다 많은 게놈 서열을 포착한다. 두 개의 절단(cut) 위치가 있는 유전자좌를 타겟팅하는 모든 방법에 대해 역방향 리드는 두 번째 컷 사이트에서 시작하므로 이러한 리드는 각 궤적에 대한 게놈의 동일한 위치에 정렬(align)하게 된다.
대조적으로, paired-end 시퀀싱을 이용한 RAD-Seq 프로토콜은 매우 다른 유형의 데이터를 생성한다. 순방향 리드는 절단 사이트에서 시작하지만 역방향 리드는 무작위로 절단된 끝에서 시작한다(일반적으로 400-700 bp). 따라서, 주어진 유전자좌에서의 역방향 리드는 길이가 엇갈리게 되며, 이들 데이터는 긴 컨티그(contig)를 어셈블하는데 사용될 수 있다. 예를 들어, 라이브러리 조각의 길이 조정이 된 경우 contig는 1 kb까지 길어질 수 있다. 이러한 RAD contigs는 paralogue의 확인을 향상시키고, 기능적으로 중요한 유전자좌의 BLAST 검색을 위해 더 많은 서열을 제공하며 계통학적 또는 계통 발생 분석을 위한 haplotype 데이터를 제공할 수 있다. 더 긴 contig 서열은 또한 PCR 프라이머 또는 서열 포착 프로브(sequence capture probe)의 디자인을 가능하게 하여 추가 연구를 위한 관심 대상 유전자좌를 타겟할 수 있다.
모든 방법에 대해, paired-end 시퀀싱으로 생성된 리드 쌍은 리드 길이와 단편 크기 범위에 따라 겹칠 수 있으므로 조각이 200-300 bp 미만이면(예: common cutter로 GBS를 사용하여 생성된 일부 단편 효소)의 경우, 리드 길이를 늘리거나 쌍으로 된 서열을 사용하면 게놈 서열 정보를 얻지 못할 수도 있습니다. 그러나 중복 리드 쌍을 사용하여 리드의 끝 부분에 대한 유전자형 정확도를 향상시킬 수 있습니다.
? 생물 정보학 분석
사후 시퀀싱 분석은 일반적으로 모든 RAD-Seq 방법을 사용하여 생성된 데이터에 대한 몇 가지 기본 단계를 공유한다. 초기 분석에는 바코드(존재하는 경우)의 demultiplexing 및 트리밍 (trimming), 기대되는 제한 효소 절단 사이트 및 시퀀스 품질에 기초한 리드 필터링, 그리고 리드의 말단 quality가 낮은 경우 트리밍을 할 수 있다. 일부 RAD-Seq 방법의 경우 초기 분석 중에 PCR 복제물을 제거하여 추후 분석에 대한 유전자형 정확도를 향상시킬 수 있다(아래 참조). 참조 게놈이 이용 가능하다면, 이 참조 게놈에 대한 리드의 정렬(alignment)에 의해 유전자좌가 식별될 수 있다. 또는 각 유전자좌는 유사한 리드들을 함께 클러스터링하고 유전자좌에서 리드들 사이의 변이가 시퀀싱 에러 또는 allelic variation 중 하나를 나타내는 것으로 새롭게 어셈블리 될 수 있다. 유전자좌 식별 후 RAD-Seq을 이용하여 얻은 paired-end 리드들에 대해 긴 contigs를 생성할 수 있다. 유전자형은 최대 가능도(maximum likelihood) 또는 베이지안 접근법을 사용하여 수행할 수 있다. 최대 가능도 추정법은 특히 베이지안 접근법이 집단 수준(population-level) 대립유전자 빈도를 사용하여 유전자형에 대한 사전 확률을 설정하는 경우 베이지안 방법보다 더 높은 depth의 적용 범위를 요구할 수 있다.
RAD-Seq 데이터를 분석하기 위해 특별히 설계된 여러 프로그램(예: Stacks, pyRAD 및 UNEAK, 기타 공개적으로 사용 가능한 스크립트 및 파이프 라인)을 사용할 수 있다. Stacks에는 품질 필터링(quality filtering) 및 유전자좌 식별(reference-aligned or de novo)에서부터 유전형 분석 및 집단 유전학(population genetics) 통계 계산에 이르기까지 분석의 모든 부분을 수행할 수 있는 여러 가지 유연한 모듈이 포함되어 있다. 계통(phylogenetic) 응용 프로그램을 위해 특별히 설계된 pyRAD는 대립유전자 간의 삽입-결실(insertion-deletion) 변이를 처리할 수 있는 장점을 가지고 있으며 높은 품질 필터링과 새로운 유전자좌 식별 및 유전자형 분석을 수행하므로 더 넓은 분류학적(taxonomic) 규모의 연구에 더 적합할 수 있다. UNEAK는 GBS 데이터와 연관 매핑(association mapping)을 위한 TASSEL 파이프 라인의 일부이며 네트워크 기반 SNP 탐지 알고리즘을 사용하지만 서열 단편의 트리밍 및 de novo 유전자좌 식별을 위한 매개 변수와 같은 특정 측면에서는 다른 소프트웨어보다 유연성이 다소 떨어진다. 또한 RADseq 데이터는 품질 필터링, 참조 게놈과의 정렬 및 유전자형 분석을 위해 보다 일반적인(generic) 소프트웨어 도구를 사용하여 분석할 수 있다.
유전자형에 따라 많은 양의 누락된 데이터가 있는 유전자좌 또는 개별 샘플을 제거하려면 일반적으로 추가 필터링 단계가 권장된다. 이 단계에서 적절한 수준의 필터링은 연구 목표 및 수행할 후속 분석에 따라 다르며, 이는 누락된 데이터 및 유전자좌의 표본 크기에 대한 민감도(sensitivity)가 다양하기 때문이다. 최근 여러 연구에서 RAD-Seq 데이터 분석의 세부 사항, 특히 de novo 유전자좌 식별에 사용되는 매개 변수가 분석 결과에 상당한 영향을 미칠 수 있는 방법을 강조하고 있다. 이 작업 중 일부는 RAD-Seq 데이터에 생물 정보 도구를 적용하는 방법에 대한 명시적인 권장 사항을 제공한다. 전반적으로, 연구자들은 품질 필터링에서 유전자좌 식별 및 유전자형 분석, 결과의 민감도를 비판적으로 평가(critically evaluate)하고 연구 목표에 따라 분석을 최적화하기 위해 분석의 모든 단계에서 사용되는 매개 변수를 변경하는 것이 중요하다.
3. 오류() 및 편향(bias)의 원인
RAD-Seq 방법들은 공통적으로 모든 차세대 시퀀싱 방법에서 나타나는 시퀀싱과 유전자형에 대한 오류를 공유하며, 또한 RAD-Seq 방법마다 특이적인 몇 가지 오류 및 편향(특히 라이브러리 준비 프로토콜 및 통계 분석에 따라)이 있다.
? Allele dropout 및 null alleles
Allele dropout은 제한효소 인식 사이트에서 다형성이 발생하여 그 위치에서 DNA를 절단하지 못하는 경우에 나타난다. Null allele은 완전한 인식 사이트가 없어서 시퀀싱되지 못하는 allele을 의미한다. 특히 절단 후 조각 길이(post-digestion fragment length)가 유전자좌 세트를 줄이기 위해 크기 선택(size selection)을 사용하는 방법에 대해 선택된 크기 범위를 벗어날 수 있기 때문에, 제한효소 절단 사이트가 없으면 인접한 절단 사이트에서 유전자좌에 대한 allele dropout을 유발할 수 있다.
Allele dropout의 빈도는 제한효소 인식 부위의 누적 길이에 따라 증가하는데, 이는 더 긴 서열에서 돌연변이의 확률이 증가하기 때문이다. 또한 다형성(polymorphism)의 전반적인 수준에 따라 allele dropout이 증가하고 ddRAD에 의해 생성된 데이터가 RAD-Seq에 의해 생성된 데이터보다 더 큰 영향을 미친다는 것이 확인되었다(역자: 전자의 경우 유전자좌는 하나가 아닌 두 개의 절단 사이트에 의존하기 때문에).
Allele dropout으로 인한 유전자형 분석 오류는 게놈 다양성의 과소평가(underestimation), Fst의 과평가(overestimation), 그리고 Fst outlier test의 false positive와 false negative의 증가 등을 통해 집단 유전 통계에 편향을 일으킬 수 있다. 그러나 유효 인구수(Ne)가 크지 않으면 이러한 편향의 영향은 제한적이라고 여겨진다. Fst 편향은 데이터 세트에서 null allele 인자가 있는 유전자좌를 제거함으로써 대체로 해결될 수 있다. 이론적으로, 어떤 개체는 유전자좌에서 하나 또는 모든 복제가 결실될 수 있기 때문에, null allele을 갖는 유전자좌는 개별 표본에 대한 depth-of-coverage의 차이가 큰 차이로 식별될 수 있어야 한다. 그러나 많은 다른 요소들도 depth-of-coverage의 차이를 유발하기 때문에(아래 참조), 이것이 항상 null alleles를 신뢰할 수 있는 지표는 아니다. 그럼에도 불구하고 빈번하게 null allele를 갖는 유전자좌는 많은 필터링 기법에 의해 제거될 수 있다. Null allele가 있는 유전자좌의 제거는 편향된 Fst 추정 문제는 대부분 해결이 가능하지만, 편향된 다양성 추정(biased diversity estimates)을 보완하는데는 거의 도움이 되지 않을 수 있다. Null allele를 가진 유전자좌는 돌연변이 속도가 더 빠르고 유전적 다양성 수준이 높은 게놈 지역에서 더 자주 발생하기 때문에 데이터 세트에서 이들 유전자좌가 없다는 것은 전체 게놈 다양성을 과소평가(underestimation)하는 경향이 있다.
? PCR 중복 및 유전자형(genotyping) 오류
대부분의 차세대 시퀀싱 라이브러리 준비 프로토콜은 클론 DNA 단편(PCR 복제물)이 원래 게놈 DNA 단편(모체 단편)에서 생성되는 PCR 단계를 수행한다. PCR 과정에서 확률론적 과정(stochastic process)에 의해 특정 대립유전자가 다른 대립유전자보다 더 증폭될 수 있다. 이러한 잠재적 치우침 현상은 이형접합체가 동형접합체로 나타날 수 있거나 PCR 오류를 포함하는 대립 형질이 참(true)인 대립 형질로 나타날 수 있기 때문에 추후 분석에서 오류를 유발할 수 있다. 연구에 따르면 PCR 중복은 RAD-Seq 데이터에서 빈번하게 발생할 수 있다(예: 20-60%의 리드 들에서). 이론상으로는, PCR은 주어진 유전자좌 위에서 하나의 대립유전자를 체계적으로 선호해서는 안되며, 따라서 많은 수의 유전자좌로부터 추정된 매개 변수가 실질적으로 편향되지 않아야 한다. 그러나 outlier 검사나 친자 확인과 같이 개별 유전자좌에서 높은 유전자형 정확도를 요구하는 분석은 PCR 복제물이 존재할 경우 잘못된 결과를 초래할 수 있다.
대부분의 차세대 시퀀싱 프로토콜을 사용하여 생성된 서열 데이터의 경우, PCR 복제물은 생물정보학적 분석을 이용하여 제거하고 유전자형 정확성을 향상시킬 수 있습니다. PCR 복제물이 게놈의 동일한 위치에서 시작하고 끝나는 단편으로 식별될 수 있기 때문에 기계적(mechanical) 또는 무작위(randomly) 효소 단편화 단계가 있는 프로토콜을 포함하는 분석에서 가능하다. 기계적 전단(sheering) 단계로 인해, 이 방법은 paired-end 시퀀싱을 사용하는 RAD-Seq에서 생성된 서열 데이터에서 PCR 복제물을 확인하는 데에도 사용될 수 있다. 어떤 특정 상황에서는(순방향 리드와 역방향 리드 사이의 거리가 매우 짧거나 로컬 범위가 매우 큰 경우), 우연히도 실제로 동일한 시작점과 끝점을 가진 조각을 제거하기도 한다.
최근에 개발된 또 다른 방법은 PCR 전에 부모 단편(parent fragment)을 표지하기 위해 시퀀싱 어댑터 내에서 degenerate 염기 영역을 사용하여 PCR 복제물을 확인하는 가능성을 보여주었다. 이 방법은 맞춤형으로 설계된 어댑터를 사용하는 모든 프로토콜에서 적용될 수 있다. PCR 복제물을 해결하기 위한 또 다른 방법은 Illumina PCR-free kit를 이용한 ezRAD에서처럼 라이브러리 준비의 PCR 단계를 제거하는 것이다. 그러나 PCR-free 키트는 현재 훨씬 비싸며 다른 RAD-Seq 프로토콜보다 많은 양의 게놈 DNA(1 μg)가 필요한 단점이 있다.
? 유전자좌(loci) 간의 시퀀싱 수준(Depth-of-coverage)의 차이
PCR 복제물과 allele dropout이 RAD-Seq 유전자좌 내 특정 대립유전자의 선호적인 (preferential) 서열 결정 결과로 유전자형 오류를 야기할 수 있는 반면에, 다른 여러 현상은 특정 유전자좌가 다른 유전자좌에 비해서 시퀀싱이 선호되는 현상을 일으킬 수 있다(역자: 유전자좌 간 시퀀싱 불균형을 의미함). 이러한 현상은 유전자형 오류를 일으키지는 않지만, 시퀀싱 빈도가 낮은 유전자좌에 대한 정확도를 얻기 위해서는 더 많은 시퀀싱 수준(depth-of-coverage)이 요구된다. 이러한 현상의 예로는 PCR 동안 GC 함량에 기반한 시퀀싱 단편의 불균등한 증폭이 있으며, 이 편향은 PCR 단계를 포함하는 모든 RAD-Seq 방법에 영향을 미친다. 또 다른 현상은 더 긴 단편에 걸친 더 짧은 단편의 특이적 증폭이다. 이 문제는 각 유전자좌위가 개별적인 조각 길이를 가지고 있기 때문에 두 컷 사이트가 위치하는 모든 RAD-Seq 메서드에 영향을 미치게 된다.
4. RAD-Seq 연구 설계 방법
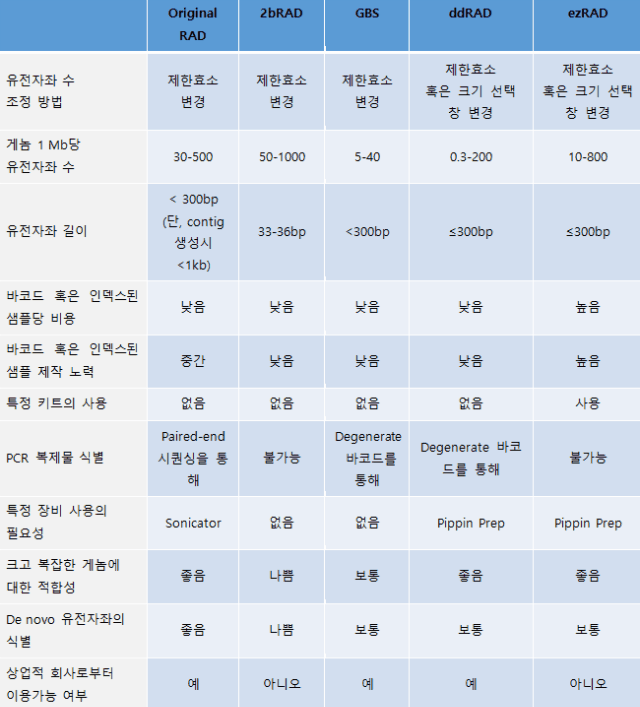
특정 애플리케이션에 대한 RAD-Seq 연구를 설계하려면 가장 적절한 RAD-Seq 방법, 샘플링 및 시퀀싱 전략, 예산 및 기타 방법론적 세부 사항과 관련하여 몇 가지 주요 사항들을 고려해야 한다(선택된 방법 간의 고려해야 할 사항 등이 표 1에 요약됨).
? 유전자좌(loci)의 수
RAD-Seq 방법으로 확인되고 유전자형이 결정되는 유전자좌의 수는 게놈 크기, 게놈의 제한효소 절단 부위의 빈도 및 염기 서열 분석 대상인 절단 부위의 수에 따라 달라진다. 계산(computational) 도구는 각 프로토콜에 대해 예상되는 유전자좌의 수를 추정하는데 사용할 수 있다. 모든 절단 부위(원래 RAD 및 2bRAD)를 대상으로 하거나 직접 크기 선택 단계(GBS)없이 일반 커터 효소를 사용하는 RADseq 방법은 일반적으로 더 많은 loci를 제공하지만 효소의 선택에 따라 그 수를 조정할 수 있다. 대조적으로 명시적인(explicit) 크기 선택 단계(예: ddRAD 및 ezRAD)가 포함된 프로토콜은 효소 또는 효소의 선택뿐만 아니라 선택한 크기 범위를 변경하여 유전자좌 수를 조정할 수 있으므로 일반적으로 적은 수의 유전자좌를 제공하는 유연성을 갖는다. 또는 RAD-Seq 프로토콜에서 유전자좌 수를 줄이는 또 다른 방법은, 유용한 RAD-Seq 유전자좌의 하위 집합에 대한 프로브를 직접 설계하고 이를 사용하여 선택한 유전자좌(즉, RAD 캡처)를 캡처하고 시퀀싱 하는 것이다.
유전자좌의 최적의 수는 당연하게도 연구의 목표에 따라 달라진다. 계통발생학적 관계, 지리적 집단 구조, 유전자 이동, 개체 근친 교배(혈통에 의한 동일성)와 같은 중립 또는 게놈 전반의 과정을 평가하는데 초점을 맞춘 연구는 종종 수백 또는 수천 개의 SNP- 포함 RAD-Seq 유전자좌를 필요로 한다. 대조적으로, 선택 표지(selection signatures)를 나타내는 것과 같이 전체 게놈에서 기능적으로 중요한 영역을 특징으로 하는 연구는 더 많은 표식 집합(예: 수십 또는 심지어 수십만 RAD-Seq 좌위)을 필요로 한다. 또한, RAD-Seq 유전자좌의 최적의 수는 염색체와 재조합 양상에 따른 연관 불균형(linkage disequilibrium)의 정도에 따라서도 달라진다. 예를 들어, 실험실 F2 교차 또는 최근에 혼합된 개체군은 outbreed 개체군보다 적은 유전자좌를 필요로 하지만, 많은 수의 자손 및 더 많은 표식자가 있으면 통계력이 증가할 수 있다. Outbreed 집단에서 연관지도를 작성하려면 더 많은 표식이 필요하다. 최근 및 과거의 효과적인 개체군 크기(effective population size) 및 근친 관계를 추정하기 위해 Chromosome stretches(예: 동형 접합성의 진행)에 따른 다양성 패턴을 정량화하기 위해서는 수만 개의 유전자좌가 필요하다.
일부 생물학적 요인들로 인해 표적화되어야 하는 유전자좌의 수가 증가될 수 있다. 예를 들어 게놈 변이가 낮은 병목 현상을 겪은 집단은 변이 수준을 정확히 정량화하기 위해 더 많은 유전자좌의 시퀀싱을 요구할 수 있다.
? 시퀀싱 리드(read)의 유형
긴 리드 또는 paired-end 리드는 분석에 많은 이점들을 제공한다(유전자좌 식별, paralogous 서열 또는 반복 서열의 식별, 기능적으로 중요한 유전자좌를 검색하는 BLAST 분석 등). 대부분의 RAD-Seq 프로토콜의 경우, 서열 길이는 시퀀싱 기술에 의해 주로 결정된다(역자: 일반적으로 Illu-mina를 사용하며 최대 150 bp까지를 생산하지만, 일부 경우에는 300 bp까지도 가능함). 상대적으로 짧은 단편(예: 100 bp) 및 single-end 시퀀싱으로 많은 연구 문제를 충분히 해결할 수 있다. 그러나, 위에서 설명한 것처럼 paired-end 시퀀싱을 이용한 RAD로부터 contig를 조립함으로써 더 긴 RAD-Seq 유전자좌를 얻을 수 있으며, 이 방법은 참조 게놈이 없는 복잡한 게놈에 특히 유리할 수 있다. 모든 방법 중에서 2bRAD는 가장 짧은 단편(33-36 bp)를 생성하므로 이 기술은 de novo 유전자좌 식별 또는 크고 복잡한 게놈(예: 인간 게놈)의 경우에는 권장되지 않는다.
? 이미 제공된 게놈 자원(prior genomic resources)
사전 참조 서열은 RAD-Seq 연구에 많은 이점을 제공할 수 있다. 참조 게놈 서열뿐만 아니라 낮은 품질로 조립된 게놈 스케폴드(scaffold) 또는 심지어 이전 연구에서 제공된 RAD 유전자좌 세트들을 이용해서 paralogous 또는 반복 서열을 필터링하고 삽입-삭제 변이를 확인하며 비-표적 DNA 서열(박테리아 서열 등)을 제거하는 등 분석 능력을 크게 향상시킬 수 있다. 또한 잘 구축된 참조 서열을 이용하여, 유전자좌의 물리적 위치에 대한 정보를 사용하여 여러 유전자좌를 커버하는 더 큰 염색체 지역 전체에 걸쳐 haplotype을 추론하는 매핑 연구도 수행될 수 있다. GBS 및 MSG 방법은 부모 조상(parental ancestry)의 염색체 블록(chromosomal blocks)이 상당히 큰 모델 종에서 이러한 방식을 이용하여 형질지도 작성을 사용하였다. 집단 게놈 연구는 참조 게놈 어셈블리를 사용하여 슬라이딩 윈도우 분석을 수행할 수 있게 되었고, 또한 집단 간의 분기 선택(divergent selection) 하에 있는 영역과 같이 관심 있는 게놈 영역을 탐지하는 통계적 능력을 높이는 데도 사용될 수 있다. 또한 참조 게놈이 없는 경우 RAD-Seq 프로토콜로 생성된 긴 contigs는 paralogous 또는 반복 시퀀스를 구분할 수 있는 정보를 제공한다.
? 시퀀싱 수준(depth of sequencing coverage)
모든 RADseq 방법의 라이브러리는 서로 다른 수준의 양으로 시퀀싱이 될 수 있으며 가장 이상적인 시퀀싱 양은 수행하는 연구에 따라 다르다. 극단적인 경우, 잘 구축된 참조 게놈을 이용한 연구에서는 매우 적은 양의 시퀀싱으로도 효율적인 연구가 가능하다. 참조 유전체가 없는 경우 훨씬 더 많은 시퀀싱 양이 필요하다(예: 10-20배). 만약 개별적인 샘플을 모두 모아서 de novo 어셈블리를 하는 경우에는 이보다 적은 시퀀싱 양(예: 5배)으로도 가능하다(하지만 이 경우엔 유전자형을 결정하기 전 시퀀싱 단편들을 각 샘플에 따라 분리시켜주는 작업이 선행되어야 한다). 배수체를 가진 taxa에서는 더 많은 양의 게놈 커러리지가 요구된다(같은 양의 서열 단편당 haploid 게놈 커버리지의 값이 줄어들기 때문에).
? 연구 예산
RADseq 데이터를 생성하는데 쓰이는 상당 부분의 비용은 시퀀싱에 소모된다. 시퀀싱을 하기 위해서는 유전자좌 수, 표본 수 및 집단, 개체당 유전자좌 수 등을 고려해야 한다. 그러나 서로 다른 프로토콜은 라이브러리 준비 비용과 샘플 준비 횟수에 따라 준비 비용이 크게 달라질 수 있으므로 더욱 신중한 선택이 요구된다. 예를 들어 original RAD-Seq 프로토콜은 상대적으로 많은 단계를 가지고 있기는 하지만 프로토콜 초기 단계부터 멀티플렉싱이 가능하기 때문에 샘플의 증가에 크게 비용이 증가하지 않았다. 하지만 ezRAD의 경우 마지막 단계 전까진 멀티플렉싱을 적용할 수 없기 때문에 샘플에 비례해서 비용이 올라감으로 이 방법은 소수의 샘플 또는 샘플 풀에 가장 적합할 수 있다. 일부 RADseq 프로토콜은 또한 특수한 바코딩을 필요로 하는 어댑터 때문에 초기에 많은 비용이 들기도 하며 특수 실험 장비 구입을 요구할 수 있다. Original RAD-Seq는 DNA sonicator가 필요하며 직접 크기 선택 단계(예: ddRAD 및 ezRAD)를 사용하는 RAD-Seq 프로토콜은 Pippin Prep을 이용하여 크기 선택의 정확성과 일관성을 높일 수 있으며 교차 오염의 가능성을 줄일 수 있다.
? 데이터의 비교(가능)성(compatability)
RADseq 연구를 설계할 때 최종 고려 사항에는 시퀀싱 작업 전반 및 실험실 전반에 걸친 데이터의 일관성이 있다. 크기 선택이 일관되지 않으면 유전자좌 세트를 줄이기 위해 크기 선택을 사용하는 방법에서 라이브러리마다 차이를 야기하는 등의 문제가 생길 수 있다. 여러 크기 선택 기술(자동 또는 수동 젤 추출과 비드 기반 선택)의 일관성에 대한 정확한 벤치마킹은 아직 수행되지 않았으나 마그네틱 비드를 이용한 것은 일관성 면에서 굉장히 떨어진다. 모든 절단 사이트(original RAD 및 2bRAD)를 대상으로 하는 방법은 일반적으로 라이브러리 간에 일관성이 높을 것으로 예상되지만 다른 잠재적 오류 유발 원인들을 포함한다(위에서 언급하였음).
? 대체 또는 보완적인 접근법
RAD-Seq은 SNP 유전형 분석 및 발견을 위한 도구로써 많은 이점을 가지고 있지만, 모든 생태 및 진화 연구를 위한 최상의 선택 방법은 아니다. Tranome sequencing (RNA-seq)과 표적화(probe-based) capture는 차세대 시퀀싱을 이용하는 두 가지 주요 대안적 축소 표현 접근법이다. 또한 전체 게놈 및 리시퀀싱 그리고 전체 게놈 풀시퀀싱 등은 표현 기법을 축소하는 것보다 훨씬 많은 게놈 정보를 제공한다. 그러나 집단 연구를 위한 전체 게놈 리시퀀싱의 가능성이 증가하고 있음에도 불구하고 많은 생태학적 진화론적인 질문은 게놈 차원의 데이터 증가로 인해 거의 해결되지 못하고 있다. 대립유전자 빈도 또는 연쇄 불균형에 기반한 선택을 탐지하기 위해 수십만 개의 마커를 사용하는 RADseq 연구에서도 마커의 밀도보다는 샘플링 수에 의한 제한점이 더 크기 때문이다.
다른 게놈 접근법들은 특정 시스템에서 보다 포괄적이거나 유연한 조사를 위해 RAD-Seq를 보완하는데 사용될 수 있다. 예를 들어, 시퀀싱 및 어셈블리 기술이 지속적으로 향상됨에 따라 비모델 생물종에 대한 de novo reference genomes의 개발이 점차 가능해지고 있으며 이러한 reference는 population-level sampling에서 추출한 RAD-Seq 데이터 분석에 많은 이점을 제공한다. 전사체 시퀀싱은 코딩(및 아마도 기능적) 서열을 목표로 하여 RAD-Seq 데이터를 보완할 수 있다. 또한 RAD-Seq은 중요한 유전자좌에 집중하기 위한 대규모 연구의 첫 번째 단계로 사용될 수도 있다. 예를 들어, RADseq는 관심 대상 후보 유전자를 확인하기 위해 게놈 전체 스캔을 제공할 수 있으며, 이러한 유전자좌의 서열 데이터를 사용하여 시퀀스 캡처를 위한 프로브를 설계할 수 있다. 그 후, 샘플당 비용을 대폭 절감된 품질이 좋지 않은 DNA를 사용하여 많은 수의 시료에 대해 차후의 표적 서열 분석을 수행할 수 있다.
표 1. 5가지 RAD-Seq 방법의 장점과 단점 요약
5. 결론
RAD-seq 기술은 생태 및 진화 유전체학 분야에서 SNP 발견 및 유전자형 분석에 많은 이점을 가지고 있지만 연구자는 여러 가지 요소들을 신중하게 고려해야 한다. 라이브러리 준비의 기술적인 세부 사항과 비용뿐 아니라 생산된 데이터의 유형과 유전자형 오류 및 편향의 원인에 따라 수많은 RAD-Seq 프로토콜이 개발되었다. 따라서 연구 질문, 연구 시스템 및 예산에 따라 적합한 방법을 사용해야 한다. 시퀀싱 기술 및 비용의 급격한 변화에도 불구하고 RAD-Seq와 같은 축소 표현 시퀀싱 접근(reduced representation sequencing approaches) 방식은 당분간 자연 집단에 대한 유전체 연구에 중요한 도구가 될 것이다.
☞ 자세한 내용은 내용바로가기 또는 첨부파일을 이용하시기 바랍니다.