

기술동향
개인유전체(personal genome)를 활용한 질병 위험 예측의 허와 실
- 등록일2019-07-03
- 조회수6936
- 분류기술동향
-
자료발간일
2019-06-20
-
출처
한국분자·세포생물학회
- 원문링크
-
키워드
#개인유전체#정밀의료
개인유전체(personal genome)를 활용한 질병 위험 예측의 허와 실
김정수 조선대학교 의생명과학과
[목차]
1. 들어가며.
2. 집단유전학의 서막
3. 질병의 진화유전과 복합유전질병(Complex disease)
4. 인간게놈프로젝트와 HapMap프로젝트, 그리고 전장유전체연관성연구(GWAS)
5. 질병의 유전이질성(genetic heterogeneity)과 다인자성(polygenicity)
6. 전장유전체연관성연구(GWAS)를 넘어
7. 개인유전체를 활용한 개인정밀의료를 향해
들어가며.
2018년 Batten 질병을 앓게 된 6살짜리 소녀 Mila를 구하자는 내용의 “Saving Mila” 클라우드 펀딩이 미국 내에서 화제였다[1]. 소녀의 안타까운 소식도 소식이었지만 병의 진단과 병의 원인이 된 희귀유전변이 탐색과정, 그리고 Mila만을 위한 맞춤형 약인 milasen의 개발과 미국 식약처(FDA)의 임상허용까지, 완벽한 개인 맞춤형 의료의 훌륭한 예가 되었기에 지금까지도 회자되고 있다[2].
‘4745일, 3,000,000,000달러’ 불과 20여년 전 한 사람의 게놈(유전체) 전체를 읽어내는데 필요했던 시간과 비용이다. 반도체 개발 속도인 무어의 법칙1 과 종종 비교되고 있는 게놈 해독 기술의 발전 속도는 가파르게 가속화되어 지금은 시중에서 누구나 약 1,000달러의 비용을 지불하면 3~4일 안에 충분히 자신의 게놈프로젝트를 성공적으로 수행할 수 있다. 이러한 속도에 더해 개인 게놈정보를 바탕으로 건강 상의 위험을 평가하기 위한 유전자 건강위험 검사(Genomic Health Risk Test) 서비스에 대한 기업들의 수요와 최근 미국 FDA의 규제 간소화에 대한 설명 발표[3]는 관련분야를 가가속(Jerk)화 하고 있다.
그러나 질병 유전체에 대한 실체적 이해의 깊이와 의료계 각 분야에서의 활용 가능성에 대한 현실은 어떠한가? 답부터 말하자면, 질병유전체 분야는 필자가 생각했던 것 보다 훨씬, 아니 굉장히 엄밀하다는 것과 그래서 임상 적용이 가능한 수준이긴 하나 아직도 가야 할 길이 많이 남았다는 다소 뻔한 답이다. 조금 더 관심있는 독자를 위해 짧지만 이러한 뻔한 답에 도달하게 된 과정을 필자가 생각하는 현재 질병유전체 연구의 허와 실을 바탕으로 살펴보고자 한다.
집단유전학의 서막
집단유전학(population genetics)은 집단에서 나타나는 유전형질에 대한 분포와 조성 변화 등을 유전과 진화의 관점을 이어 연구하는 분야이다. 집단유전학의 시작점을 놓고 1865년 Gregor Mendel의 이산형질(discrete phenotype)의 유전법칙 발견[4]을, 혹은 1886년 Francis Galton의 회귀모형을 이용한 연속 형질(continuous phenotype)에 대한 연구[5]을 언급하기도 하나 보통은 이 두 계파의 오랜 분쟁을 잠재우는, 멘델유전학과 다윈진화론의 수리적 통합 모델을 제시한 1918년 Ronald Fisher의 기념비적 논문[6]을 그 시작으로 본다.
유전(inheritance) 혹은 유전력(heritability)의 개념은 유전적 요인에 의한 형질의 변화에 대한 설명력을 말하는데 아이러니하게도 연속 형질 유전에 대한 개념으로 확장된 진화론의 창시자인 Charles Darwin은 진화를 후천적 형질로 인식했기 때문에 진화와 유전과의 연관성은 멘델에 의해 시작됐다고 보는 것이 일반적이다. 그러나 멘델의 유전법칙은 이산형질의 유전현상은 성공적으로 설명할 수 있었으나 키와 같은 연속형 형질에 대한 유전력 연구결과를 설명하는데 한계가 있었고 이를 비롯한 유전학의 많은 문제들을 설명하기 위한 수리통계학적 이론들이 만들어지고 Biometrica와 같은 저널이 출판되면서 지금의 통계유전학의 모태가 된다. 그 중 가장 핵심적인 내용이 바로 다인자 유전(polygenic inheritance)에 대한 Fisher의 통찰인데 다인자 유전의 도입은 이산형질의 유전을 설명하는 멘델의 법칙으로부터 연속형 형질의 설명이 가능2하다는 것을 수리적으로 보이면서 진화유전학에 수학적 엄밀성과 체계성을 불러오게 된다.
질병의 진화유전과 복합유전질병(Complex disease)
집단유전학의 세부연구분야로 질병을 관심 형질로 하여 질병의 유전과 진화적 원리를 설명하고자 한 질병유전학 역시 이러한 수리적 체계화를 바탕으로 발전하여 지금의 전장유전체연관성연구(Genome-Wide Association Study, GWAS)에 이르기까지 그 명맥을 이어오고 있다. 간단히 말해 질병과 연관된 유전변이가 집단에서 도태되지 않고 계속해서 유전자 풀에서 살아남아 진화되는 원리를 설명하기 위해서 de novo (or rare) variant의 발생을 비롯한 생물학적 현상과 함께 selection-drift 균형 등 다양한 진화적 개념의 도입, 그리고 이를 설명하기 위한 복잡하고 엄밀한 수학적 모델들이 한데 어우러져 지금의 질병유전3 연구가 진행되고 있는 셈이다.
글 서두에 소개한 Mila의 희귀질병(rare disease)과는 다르게 보통의 질병(common disease)들은 복합질병(complex disease), 즉 여러 유전변이들과 또 유전 외적인 요인들이 복합적으로 작용하여 발생하는 질병이다. 뒤에 그 차이를 좀 더 다루게 될 복합질병의 특성인 질병의 유전이질성(genetic heterogeneity)[7]과 다인자성(polygenicity)[8]의 존재로 인해 그 원인을 찾는 것이 간단치 않다. GWAS 분석이 활발해지기 전까지는 가설에 기반한 특정 유전자와 질병의 연관성 연구가 일반적이었다. 그러나 2002년 Hirschhorn 그룹에서 발표한 논문[9]에서 가설기반 유전질병 원인유전자 발굴의 한계점이 명백히 드러남과 동시에 게놈 프로젝트와 HapMap 프로젝트, 그리고 마이크로어레이 기술의 상용화에 힘입어 복합질병연구의 프레임이 바뀌게 된다.
인간게놈프로젝트와 HapMap 프로젝트, 그리고 전장유전체연관성연구(GWAS)
노이즈 마케팅이 있었던 것이 사실이나, 인간게놈프로젝트(Human Genome Project, HGP)는 인간의 DNA염기서열을 모두 해독하고, 이어지는 HapMap 프로젝트를 통해 반수체(haplotype) 지도를 완성하는 과정에서 3만여 개의 유전자 동정 및 위치 확인, 유전체에 존재하는 10,000,000여 개 이상의 단일염기서열상동성(Single Nucleotide Polymorphism, SNP) 존재 확인 등 여러 난치성 질환의 원인 유전자 진단과 치료법 개발의 길을 열렸다. 그러나 통계학적 관점에서 보자면 수많은 SNP의 존재는 이른 바 다중비교의 문제(multiple comparison problem)4로 인해 좋은 일만은 아니다. 너무 많은 정보는 잡음과 실제 신호를 구분해 내기가 어렵게 만든다.
다행히 이 문제를 완화시켜주는 것은 Linkage Disequilibrium 블락(block)의 존재인데, 결국 물리적으로 가까이 위치한 SNP들이 블락 단위로 함께 유전되는 경향이 있기 때문에 모든 변이를 관찰할 필요없이 LD 블락을 잘 나누고 이 블락을 대표할 수 있는(tagging) SNP들만 확인하면 전체 SNP을 모두 살펴보지 않아도 된다는 것이다. 전장유전체 정보를 최대한 많이 담을 수 있도록 tagging SNP들을 잘 선별하여 마이크로어레이로 제작하고 이를 질병-대조군(case-control) 연구디자인에 적용한 일련의 연구들은 2005년 Science지에 게재된 Age-Related Macular Degeneration 연관 유전변이 발굴[10]을 시작으로 지금까지 무수히 많은 질병연관 유전변이들이 Nature genetics 혹은 관련 저널에 보고하고 있다. 50만여 개의 tagging SNP으로 univariate analysis를 수행 후, 분석된 수만큼의 다중비교문제를 보정하기 위해 그 어느 분야보다도 엄밀한 수준의 통계적 유의성을 기준(genome-wide significance level, 10-8)을 적용하는 분석연구가 GWAS의 대명사로 수행되고 있다.
그러나 이러한 (특별한 형태의) GWAS는 한계를 갖는다. 검정한 결과가 tagging SNP을 이용한 것이기 때문에 갖는 해석의 한계부터 여전히 너무 많은(106) SNP에 비해 턱없이 적은 표본수를 분석하기 위해 부득이 개별 SNP의 제한된 효과(marginal effect)를 볼 수 밖에 없는 분석방법에 이르기까지..., 여기서 그 많은 문제들은 모두 다룰 수는 없으나 소위 ‘사라진 유전력(missing heritability)’으로 알려진 문제 제기[11]는 익숙해져 가고 있던 GWAS에 새로운 방향의 필요성을 요구하게 된다. 사라진 유전력의 문제는 간단히 말해 GWAS로 밝혀진 유전변이(혹은 tagging SNP전체)들이 알려진 형질의 유전력 중 일부 밖에 설명하지 못하는 것을 말하는데 동일한 상황이 질병유전학이 풀어야 할 숙제로 남아있다.
질병의 유전이질성(genetic heterogeneity)과 다인자성(polygenicity)
Mila의 사례에서 본 Batten질병과 같은 희귀질병(rare disease)의 경우 그 가족의 유전체를 함께 활용한 linkage 분석을 통해 그 병인규명이 어렵지 않게 이루고 있다. 그것이 가능한 것은 질병이 Mendelian 유전을 따른다는 것과 특정유전변이가 질병에 강력히(다른 요소에 영향 받지 않고) 작용한다는 점 때문이다. 암, 제2형당뇨, 그리고 알츠하이머병과 같은 보통의 질병(common complex disease)은 아직 넘어야 큰 산이 우리 앞에 버티고 있다.
유전성 난청(nonsyndromic hearing loss, NSHL)은 멘델의 유전규칙을 따르는 유전질환으로 NSHL의 원인유전변이의 존재가 형질유전에 치명적이지 않기 때문에 지금까지 도태되지 않고 되고 이어져 내려올 수 있었다. 이 경우 가족이 유전체정보를 활용하면 특정 병인규명이 어렵지 않게 가능한데 필자가 분석했던 가족의 경우 ACTG1 유전자에 존재하는 유전변이가 그 원인이었다[12]. 그런데 NSHL을 가진 또 다른 환자들을 분석한 경우, 이와는 전혀 다른 유전자에서 그 원인이 밝혀져왔다[13]. 즉, NSHL을 일으키는 유전자(혹은 유전변이)는 하나가 아니었던 것이다. 지금까지 밝혀진 NSHL유발 유전자는 90개가 넘으며 동일 유전자라고 하더라도 원인이 된 유전변이가 다른 경우도 존재한다[14]. 이러한 질병유발 유전원인의 이질성, 즉 질병의 유전이질성(genetic heterogeneity)이 문제 해결을 어렵게 하는 원인 중 하나이다.
또 다른 예를 살펴보자. 치매(dementia)증상을 보이는 사람들의 경우 그 원인을 살펴보면 대략 7~80%정도는 알츠하이머병에 기인하고 그 외 혈관성 원인 등 다른 질병에 기인하는 것이 알려져 있다. 치매 증상의 가장 많은 비중을 차지하는 알츠하이머병(Alzheimer’s disease, AD)은 아밀로이드베타(Aβ) 단백질의 비정상적 축적으로 야기되는 뇌신경퇴화를 말한다[15]. 비정상적 Aβ 누적의 이유는 사람마다 다를 수 있다. AD발병에 치명적인 원인이 되는 유전변이를 갖고 태어나는 경우를 Early Onset AD, 혹은 EOAD라고 말하며 그 발병시기가 빠를수록 그 윈인유전변이는 유전되어 왔을 가능성이 낮아진다. 그러나 보통, AD는 그 발병시기가 늦기 때문에 Late Onset AD, 줄여서 LOAD라고 부르며, 이 경우 질병 발병에 치명적이지 않은 다양한 유전변이(small effect size)들이 진화의 과정에서 살아남아 질병을 발생하게 하는 것이다. 그림 1에서 보는 것과 같이 유전정보가 형질에 영향을 주는 과정에서 같은 질병이더라도 그 밑단에서는 서로 다른 변이들이 복잡한 관계를 통해 발병의 원인이 될 수 있으며 이것을 질병의 다인자성(polygenicity)라고 말한다. 보통의 복잡질환(common complex disease)의 경우, 유전적 이질성(genetic heterogeneity)에 더해 다인자성이 모두 관여할 가능성이 높기 때문에 이것을 모두 고려한 문제 해결은 간단치 않다. 사실 이 문제는 100년 전 Fisher가 이산형 형질유전의 법칙을 연속형 형질유전의 현상으로 설명하기 위해 고민했던 문제와 맞닿아 있다.
전장유전체연관성연구(GWAS)를 넘어
필자를 비롯한 많은 연구자들이 이 문제의 여러 해법을 찾고 있는데 그 중 주목할 만한 것이 바로 omni5-genic 모델이다[16]. 유전학의 핵심은 유전요소(SNP)가 형질(질병)과 어떻게 연결되어 있는지를 밝히는 것이다. 이 모델은 복합형질(complex trait)의 경우 형질과 연관된 유전변이 혹은 이들이 놓여있는 핵심 pathway 뿐 아니라 이 밖에 존재하는 다양한 연결고리를 갖는 유전자들에 의해서도 설명된다는 것이다. 문자 그대로 ‘omnigenic’ 모델이다. 사실 이러한 시도는 시스템생물학(systems biology)란 이름으로 혹은 통합오믹스모델(integrated omics model)이란 이름으로 다양하게 시도되어 왔다. 핵심은 이런 가정을 통해 다양한 omics level 간의 연결고리를 찾고 이를 토대로 유전의 개념을 확장시켜 나가야 한다는 것이다(그림 1). 이에 대한 한 구체적 예로 올해 5월에 유전자 발현의 trans 효과를 통해 omnigenic inheritance를 설명하는 통계적모델이 발표되었다[17].
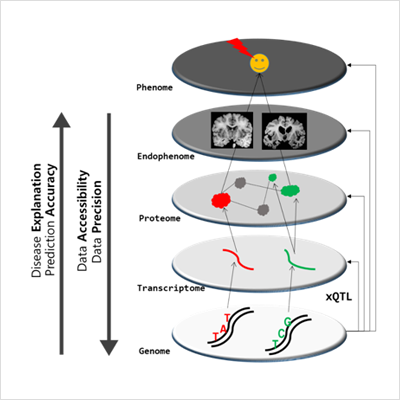
그림 1. 다양한 형태의 오믹스 수준과 수준 사이의 분석 예
개인유전체를 활용한 개인정밀의료를 향해
질병과 연관된 유전요소를 밝히려는 시도는 생각보다 굉장히 엄격한 통계적 기준과 복잡하고 잘 짜인 수리적 모델링을 통해 계속해서 발전하고 있다. 질병연관 유전요소를 찾는 궁극적인 목적은 단순한 유전형질에 대한 이해를 넘어 질병의 발병위험예측, 진단, 치료와 같은 임상적 응용에 있다. 개인유전체를 활용한 임상에의 적용, 즉 개인정밀의료는 미래의 이야기가 아니다. Mila의 고무적인 사례뿐만 아니라 암의 맞춤형진단과 치료를 위한 Cancer panel의 개발, 한정적이기는 하나 APOE유전자와 근처 유전변이를 활용한 알츠하이머병 위험예측 등, 이제는 Ancestry 분석과 같은 Entertainment 수준을 넘어 ‘엄근진6’ 해야하는 생로병사의 전단계에서 예측, 진단, 치료의 전임상단계에서 개인유전체가 활용되기 시작했다. 개인적으로는 아직 출판 전이긴 하나 연구결과들 중 제법 정확한 위험 예측도를 보이는 질병들도 있다.
물론 일부 질병의 경우 개인유전체의 활용이 이미 실용화 단계에 있지만 보다 많은 질병을 해결하기 위해서는 아직 넘어야 하는 산이 많다. 이론적 분석방법의 고도화뿐 아니라 개인유전체와의 조화를 위한 식이습관, 평소생활방식과 같은 lifelog 빅데이터의 체계적 수집에 필요한 문제들, 개인연구자수준에서 하기 어려운 산들도 존재한다. 그럼에도 불구하고 필자는 개인유전체 활용의 임상적 효용성과 실용성에 대해 낙관하고 있다. 시쳇말로 유전체연구를 직접 하는 입장에서 가성비가 제법 나오는 상황이라고 보고 있다는 것이다. 그러나 흥정은 거래의 미덕이라고 했던가? 말 나온 김에 오늘 시퀀싱 대행회사 몇 곳에 부모님 그리고 내 시퀀싱 비용을 문의해 봐야겠다.
[1] 반도체 집적회로의 성능이 24개월마다 2배로 증가한다는 경험적 법칙으로 인텔의 공동 설립자인 Gordon E. Moore가 1965년에 발표
[2] 통계학의 핵심 이론인 중심극한정리(Central Limit Theorem)과 관련된 사례로 이항분포(이산형질)의 적절한 조합이 정규분포
(연속형질)을 만들어낼 수 있음을 보임
[3] Somatic mutation에 의한 질병(예를 들어 암)이나 감염에 의한 질병과는 차이가 있음
[4] 다수의 집단 가설(a set of inferences)을 동시에 검정하는 경우 검정의 수 만큼 제 1종의 오류 발생이 증가하는 문제
[5] Omni-는 모든 것을 뜻하는 라틴어를 의미하고 이제는 익숙해진 오믹스(omics)도 같은 어원에서 유래한 단어임
[6] 엄격 진지 근엄
☞ 자세한 내용은 내용바로가기 또는 첨부파일을 이용하시기 바랍니다.
-
이전글
- IMMUNOLOGY 2019 참관기
-
다음글
- 이론 기반 촉매 설계의 동향
지식
- BioINwatch 전 세계 바이오 연구 커뮤니티를 위한 강력한 리소스, 영국 바이오뱅크(UK Biobank) 2024-06-20
- BioINpro [3D 바이오프린팅 기술] 인공지능 기반 3D프린팅 기술 및 생체의료분야 적용 기술동향 2024-04-19
- BioINwatch 정밀의료를 위한 새로운 접근, 기능 정밀의학(Functional precision medicine) 2024-02-27
- BioINpro [KRIBB 워킹그룹] 데이터 주도 과학 시대의 바이오 디지털 전환 2023-01-27
- BioINwatch 유전체 분석에 따른 정밀의료 프레임워크 설계 및 치료효과 향상에 관한 보고 2022-10-14