

기술동향
[Laura Soucek] Trends in the development of a clinically-viable Myc inhibitor
- 등록일2020-04-14
- 조회수4454
- 분류기술동향 > 생명 > 생명과학
-
자료발간일
2020-04-07
-
출처
국가항암신약개발사업단
- 원문링크
-
키워드
#clinically-viable Myc inhibitor
- 첨부파일
 그림8.jpg
그림8.jpg
Trends in the development of a clinically-viable Myc inhibitor
Vall d’Hebron Institute of Oncology, Jonathan R. Whitfield1 and Laura Soucek)
Myc and cancer
Myc is currently one of the “most wanted” targets in cancer therapy (1). The Myc family consists of three functionally related genes: c-myc (MYC), l-myc (MYCL), and n-myc (MYCN) (2). MYC was the first gene to be discovered in this family, almost four decades ago, due to homology with the viral gene v-myc, carried by the Avian virus Myelocytomatosis, which gave the name to the whole family. The other two members of the family, MYCL and MYCN were found expressed in lung cancer and brain development, respectively. All three proteins (from now on, Myc) fall in the category of intrinsically disordered proteins (IDPs), not having a well-defined three-dimensional structure in solution (1). However, Myc displays a basic-loop-helix-leucine zipper (bHLHLZ) domain at its C-terminus that allows it to dimerize with a similar protein called Myc-associated protein X (MAX), assuming a conformation suitable to DNA binding at transcriptional binding sites called E-boxes (Enhancer-boxes) (3). Together with MAX, Myc functions as a nuclear transcription factor that coordinates several intracellular and extracellular programs that contribute to the development and maintenance of tumors. In physiological conditions, it is finely tuned to be expressed only transiently, to allow cells to efficiently progress through the cell cycle (mainly, but not limited to, contributing to the G1-S transition). However, in cancer, Myc regulation is lost, due to increased Myc number, or alterations in Myc transcription, translation or even protein stability. In general, compared to other infamous oncogenes, Myc is not frequently found mutated in cancer, but it is instead induced and acts as an essential conduit downstream of other oncogenic signals, which all funnel through Myc in the nuclei of the cells to execute the transcriptional programs that ultimately lead to uncontrolled tumor growth (4). In this context, deregulated, tonic signaling through Myc can be as tumorigenic as elevated levels of it (5).
Myc has been shown to contribute to essentially all hallmarks of cancer: it promotes cell division, it contributes to angiogenesis and cross-talk with the tumor microenvironment (6), it is responsible for blocking the anti-tumor immune response (7) and it even confers better survival chances and resistance to many standard of care therapies (8).
Because of all these reasons, Myc is considered a particularly appealing target for cancer treatment. Nevertheless, no Myc inhibitor is available in the clinic yet and the race is on to develop one (9).
Myc as an “undruggable” target
Myc has long been considered a challenging – if not impossible – therapeutic target, mainly because of technical reasons, but also due to the preconceived notion that inhibiting it would cause severe potential side effects in normal proliferative tissues. From the technical point of view, the difficulties are associated to the intrinsically disordered nature of the protein, challenging to tackle with standard molecule approaches, lack of an enzymatic pocket and its nuclear localization (10).
However, different approaches have been used to circumvent these issues. Here, we are mentioning some of the best characterized strategies to date (for a complete review, refer to (9)).
1.Interference with Myc transcription
Direct strategy:
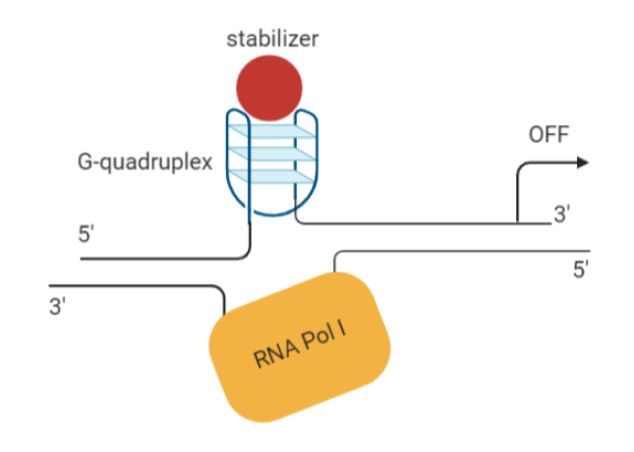
G-quadruplexes are 4-stranded DNA structures formed in guanine-rich regions. They can act as silencer elements, repressing transcription of proximal genes. The MYC promoter happens to have such a structure (11). Several studies have shown that some small molecule ligands (e.g. cationic porphyrins, quindolines) can stabilise G-quadruplexes in the MYC promoter, like CX-33543 or quarfloxin, resulting in MYC downregulation (12,13) (Figure 1). The Phase III trials for quarfloxin are currently not proceeding due to high albumin binding, but other G-quadruplexes are in the pipeline for further development (14).
Direct strategy:
Figure 1. Schematic of mechanism of action of a G-quadruplex stabilizer (created with Biorender).
Indirect strategy: BET bromodomain inhibitors.
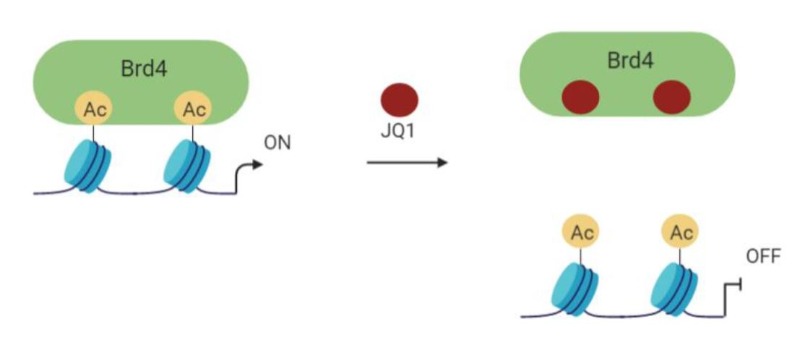
The BET bromodomain and extra-terminal domain inhibitors (BETi) have been found to be able to displace the bromodomain chromatin regulators from the large super-enhancers of genes (Figure 2). The first one to demonstrate this action against MYC as well was JQ1 in multiple myeloma, followed by Burkitt’s lymphoma and acute myeloid leukemia, where the Myc gene is amplified (15-17). It is now clear that JQ1-and other BET inhibitors- are not specific for MYC, but act on multiple genes within cancer cells that might contribute to the tumorigenic phenotype (18-20). Because of that, despite their often poor selectivity, several BET inhibitors are currently in early phase clinical trials in various malignancies (for a review, see (21).
Figure 2. Schematic of mechanism of action of a BETi such as JQ1 (created with Biorender).
2.Interference with Myc translation
Antisense Oligonucleotides (ASOs):
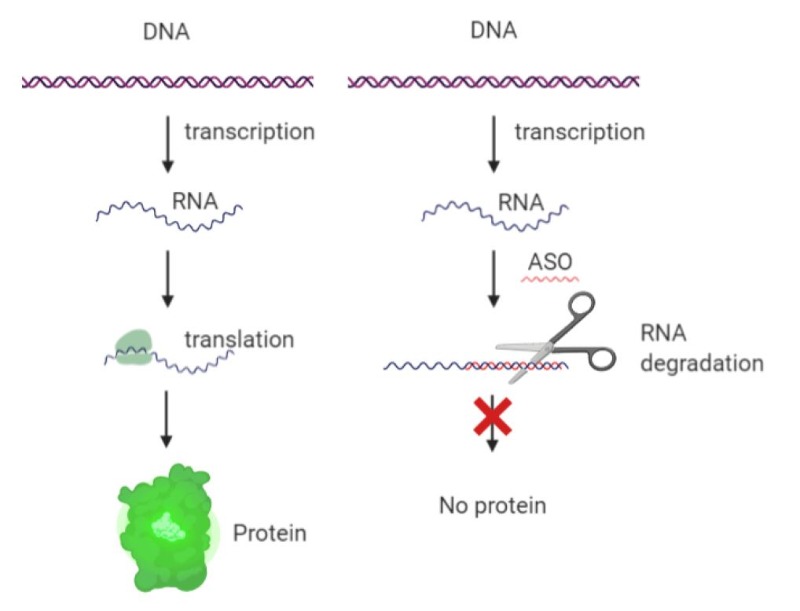
One of the first strategies tested against Myc has been the use of ASOs in order to induce degradation of Myc mRNA (22,23) (Figure 3). This approach has demonstrated some success in multiple cell lines and was tested in the clinic already two decades ago, where it got as far as Phase II clinical trials (24). Not much is reported regarding why it has not been pursued further than that.
Figure 3. Mechanism of action of ASOs (created with Biorender).
siRNA or shRNA
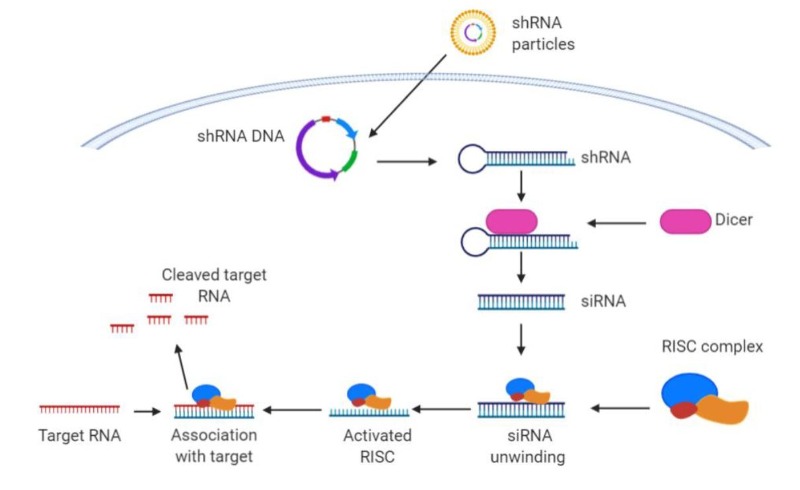
A similar approach is based on siRNA or shRNA, which takes advantage of Dicer and RISC complex to target Myc RNA before it can be translated into a functional protein (Figure 4). Once again, this approach recently reached clinical trials, where it was tested as Myc RNAi (DCR-MYC) encapsulated in lipid nanoparticles (25). Unfortunately, the trial did not meet expectations of Myc knock-down or efficacy and was discontinued.
Figure 4. Mechanism of action of shRNA and siRNA (created with Biorender).
It should be noted that, in both types of approach, a different siRNA (or shRNA) is needed for each member of the Myc family and it requires the encapsulation in nanoparticles for its delivery in vivo.
Interference with IRES-dependent translation
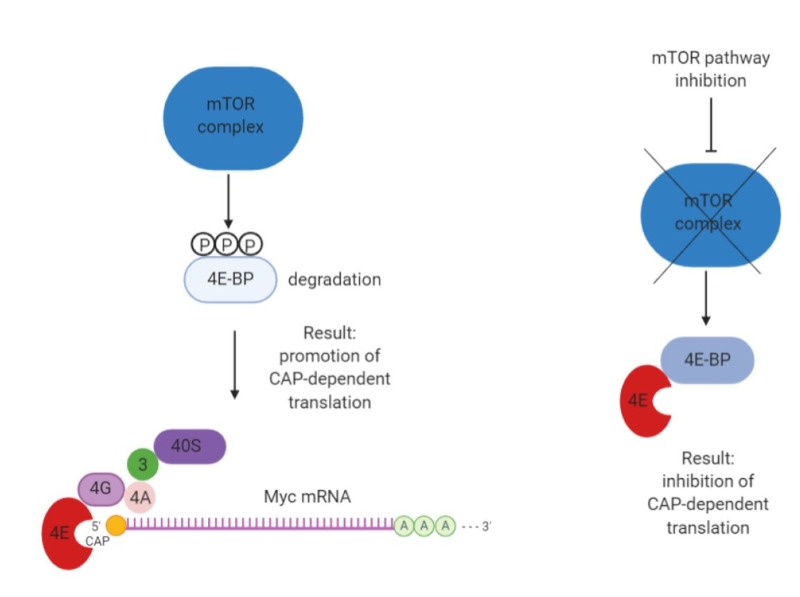
Myc mRNA can be translated both by 5’ cap- and internal ribosome entry site (IRES)- dependent mechanisms (26), offering the opportunity to regulate Myc translation by targeting this process (Figure 5).
One possibility is to inhibit mTOR itself or upstream controllers of its activity (PI3K/PTEN/AKT and Ras/Raf/MEK/ERK). In this context, a small molecule inhibitor of eIF4A, silvestrol, appeared effective in reducing Myc translation and inhibiting tumor growth (27). Multiple mTOR and mTORC1/2 kinase inhibitors are currently approved for clinical use (9), but their impact is clearly not limited to Myc alone.
Another small molecule inhibitor called saracatinib was also found to inhibit the ERK1/2-MNK1-eIF4E-mediated cap-dependent translation of Myc (28).
Figure 5. Schematic of 5’CAP-dependent Myc mRNA translation and its inhibition (created with Biorender).
3. Inhibitors of Myc dimerization and DNA binding
Small molecules
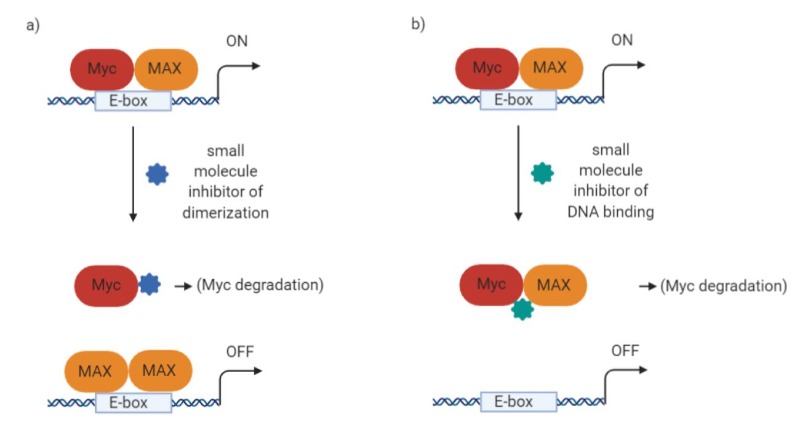
After antisense, other early attempts to inhibit Myc aimed at interfering with Myc/MAX interaction and/or preventing their binding to DNA. The first attempts were through small molecules (29,30). Most small molecules, unfortunately, have been described – at least until recently – as frequently suffering from poor bioavailability (30,31). However, more recently, small molecule inhibitors have been developed by various novel techniques and are finally starting to show better in vivo capabilities. To mention some molecules directed to disruption of Myc/MAX dimers (Figure 6a):
Small molecule 3jc48-3 is related to one of the earliest described compounds (10074-G5) and displayed more potent activity as well as a 17 hour intra-cellular half-life (32), although no further in vivo studies have been reported. Mycro3, on the other hand, was effective in vivo when administered by oral gavage, increasing survival in mouse models of pancreatic cancer (33). In addition, KJ-Pyr-9 is an inhibitor found in a Kröhnke pyridine library; it has a very low KD (6.5 nM) and blocks the growth of Myc-amplified human cancer cell line xenografts and can so penetrate the blood-brain barrier (34). Novel computational techniques are also being used to virtually screen binding to different intrinsically disordered protein conformations, and compounds were identified with micromolar affinity for MYC (35).
Among other compounds that can disrupt binding to DNA, we would like to mention KSI-3716, which was effective in mouse models of bladder cancer (36,37) (Figure 6b).
Figure 6. Schematic of the mechanism of action of a) a putative Myc/MAX dimer destabilizer; b) a potential inhibitor of Myc/MAX binding to DNA (created with Biorender).
Disappointingly, further development of these promising molecules has not been reported. Given the number of effective small molecules out there that are hampered by in vivo delivery issues, it would also be interesting to see more attempts at incorporating these compounds into nanoparticles. To this end, MI1-PD is an integrin-targeted lipid-encapsulated nanoparticle formulation of a Myc-MAX dimerization inhibitor that showed in vivo efficacy in a mouse model of multiple myeloma (38).
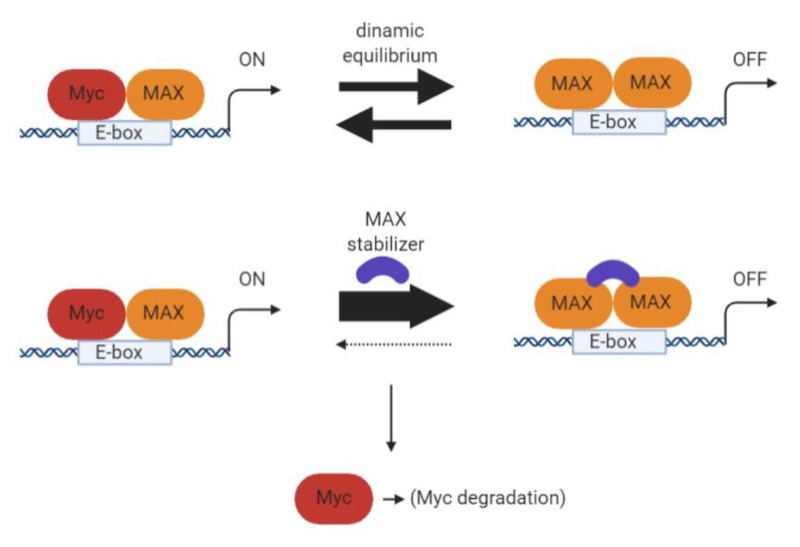
A contrasting approach would be to stabilize Myc’s binding partner MAX, thus occupying DNA target sites with MAX homodimers and preventing MYC transcriptional activity (39). Recently such compounds have been described as antagonizing MYC-dependent gene expression in cells and delaying growth of MYC-driven tumors (40) (Figure 7).
Figure 7. Schematic of the mechanism of action of a MAX/MAX dimer stabilizer (created with Biorender).
Mini-proteins or protein domains
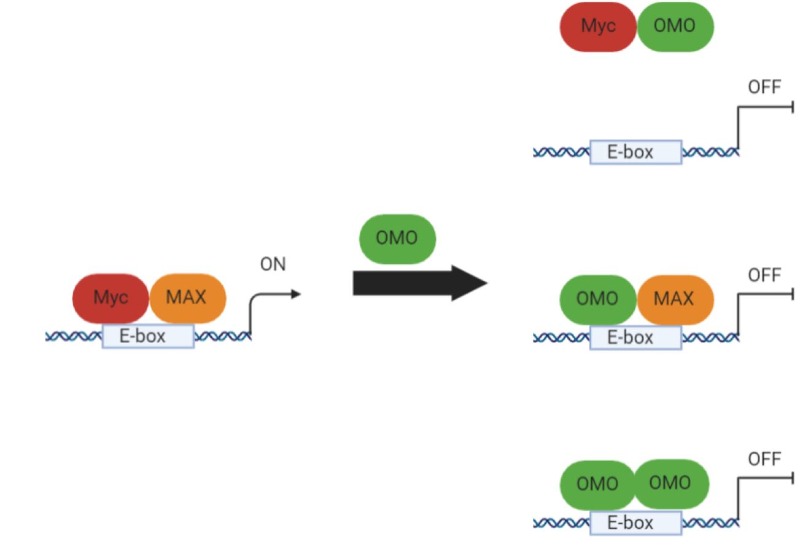
Several peptides and mini-proteins based on domains from Myc family members have been described as potential therapeutics tools. The first and most characterized is Omomyc, a mutant MYC bHLHLZ domain, functioning as a very efficient Myc dominant negative (41,42). Omomyc functions through several mechanisms of action, forming heterodimers with Myc unable to bind DNA, but also homodimers and heterodimers with MAX, which occupy E-boxes with transcriptional inactive complexes (Figure 8). Omomyc was initially used as a transgene in multiple mouse models of cancer to establish the proof-of-concept that Myc inhibition is feasible, effective and lacks significant side effects (43-49). Recently, the recombinantly-produced Omomyc mini-protein has been shown to have unexpected cell penetrating properties and therapeutic activity in NSCLC (non-small-cell lung cancer), both in vitro and in vivo, and it is being developed as a drug, expected to reach clinical trials in 2021 (10,50).
It should be noted that, based on the Omomyc basic region, some smaller peptides have been developed for interference with MYC/MAX binding to the E-box. Such peptides recapitulated at least one of Omomyc’s mechanisms of action and have shown promise in vitro, but remain to be tested in vivo (51).
Another mini-protein modulating the Myc network has been derived from MXD1: Mad. Like Omomyc, Mad dimerizes with MAX and binds the E-box, interfering with Myc-mediated transcription in cell models (52).
Figure 8. Omomyc mechanism of action as Myc dominant-negative (created with Biorender).
4.Induction of Myc degradation
In physiological conditions Myc has a short half-life (approximately 20 minutes), mainly determined by its phosphorylation and consequent ubiquitination. Hence, a potential strategy for its inhibition is to increase the activity of ubiquitinases or inhibit deubiquitinases (Figure 9).
To mention some examples of these applied strategies:
Oridonin has been associated to activation of FBW7-mediated degradation of MYC (53). Its derivatives reached clinical trials (e.g., HAO472 for leukemia treatment), however its anti-cancer function has not been linked to MYC only.
For what specifically concerns MYCN, it has been shown that complexes with Aurora-A kinase can protect it from proteasomal degradation (54), hence Aurora-A inhibitors (i.e. MLN8054 and MLN8237) have been developed to overcome this protection. Unfortunately, MLN8054 was terminated by Millenium in 2008 due to side effects (55), but the second generation inhibitor MLN8237 (Alisertib) is currently being evaluated in multiple Phase II and III studies and some positive results have been reported (56-58).
Small molecule inhibitors against another ubiquitin ligase, HUWE1, have also been shown to induce Myc and Miz1 degradation (59).
Figure 9. Schematic of induction of Myc degradation by modulation of the ubiquitin pathway (created with Biorender).
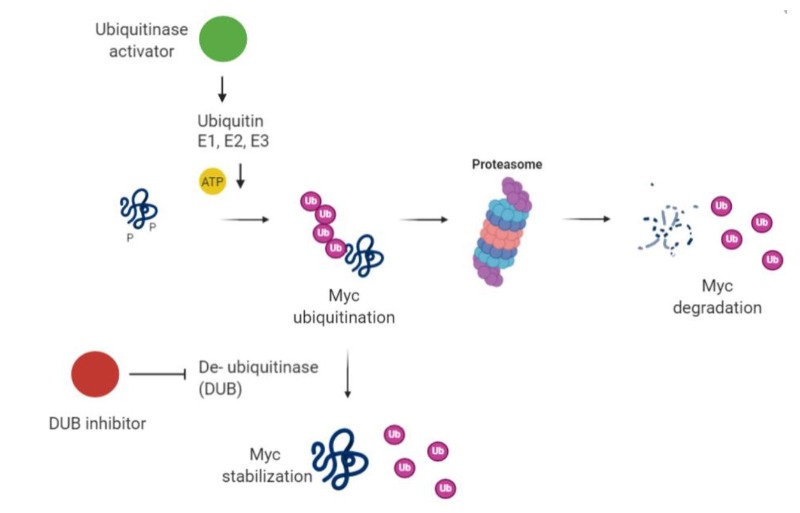
Alternatively, the tumor suppressor protein phosphatase 2A (PP2A) destabilizes MYC by targeting serine 62 of MYC (60). Cellular inhibitors of PP2A (the SET oncoprotein and CIP2A, the cancerous inhibitor of PP2A) are increased in human cancers and lead to overexpression of MYC (61). Thus, inhibitors of SET and CIP2A can reduce MYC activity (62,63) and are in preclinical development, though they would also act on other targets besides MYC alone.
A potential arrival to this field could be PROTACS – proteolysis targeting chimeras – that couple a protein-targeting ligand with an E3 ubiquitin ligase (64). This approach relies on degradation of the target upon binding. Whether this promising new approach will be applicable to a protein like Myc with such a short half-life remains to be seen, but indirect targeting with a pan-BETi PROTACS ARV-771 reduced MYC expression and caused xenograft tumor regression in prostate cancer mouse models (65). Clinical trials have not been announced as yet.
5. Synthetic lethality
In addition to the plethora of strategies already described, other indirect approaches have also been employed to get around the issues involved with direct Myc targeting. These include synthetic lethal approaches that could in theory target any specific dependency of Myc-driven tumor cells. Many diverse targets have already been identified, including SAE1/2 (66), CDK2 (67), CDK9 (68), PIM kinase (69), and many more involved in metabolism (for a more complete discussion see (9)).
Challenges and opportunities
We can all agree that this is an exciting time in the Myc inhibition world: many groups – and now also Biotechs and Big Pharma – are involved in the hunt for the first and best Myc inhibitor that can find its application in cancer patients. With so many strategies now to tackle this previously undrugged and seemingly undruggable target, it finally feels like clinical success is fast approaching. We all feel hopeful that such a historical milestone will revolutionize cancer treatment and potentially become applicable to multiple types of indications in oncology.
References
1. Dang CV, Reddy EP, Shokat KM, Soucek L. Drugging the ‘undruggable’ cancer targets. Nature reviews Cancer 2017;17(8):502-08.
2. Masso-Valles D, Beaulieu ME, Soucek L. MYC, MYCL and MYCN as therapeutic targets in lung cancer. Expert opinion on therapeutic targets 2020.
3. Tansey WP. Mammalian MYC Proteins and Cancer. New Journal of Science 2014;2014:27.
4. Dang CV. MYC on the path to cancer. Cell 2012;149(1):22-35.
5. Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, et al. Distinct thresholds govern Myc’s biological output in vivo. Cancer cell 2008;14(6):447-
57.
6. Whitfield JR, Soucek L. Tumor microenvironment: becoming sick of Myc. Cellular and molecular life sciences : CMLS 2012;69(6):931-4.
7. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016;352(6282):227-31.
8. Carabet LA, Rennie PS, Cherkasov A. Therapeutic Inhibition of Myc in Cancer. Structural Bases and Computer-Aided Drug Discovery Approaches. International
journal of molecular sciences 2018;20(1).
9. Whitfield JR, Beaulieu ME, Soucek L. Strategies to Inhibit Myc and Their Clinical Applicability. Frontiers in cell and developmental biology 2017;5:10.
10. Beaulieu ME, Soucek L. Finding MYCure. Molecular & cellular oncology 2019;6(5):e1618178.
11. Yang D, Hurley LH. Structure of the biologically relevant G-quadruplex in the c-MYC promoter. Nucleosides, nucleotides & nucleic acids 2006;25(8):951-68.
12. Brooks TA, Kendrick S, Hurley L. Making sense of G-quadruplex and i-motif functions in oncogene promoters. The FEBS journal 2010;277(17):3459-69.
13. Brown RV, Danford FL, Gokhale V, Hurley LH, Brooks TA. Demonstration that drug-targeted down-regulation of MYC in non-Hodgkins lymphoma is directly mediated
through the promoter G-quadruplex. The Journal of biological chemistry 2011;286(47):41018-27.
14. Asamitsu S, Obata S, Yu Z, Bando T, Sugiyama H. Recent Progress of Targeted G-Quadruplex-Preferred Ligands Toward Cancer Therapy. Molecules 2019;24(3).
15. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011;146(6):904-17.
16. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013;153(2):320-34.
17. Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proceedings of
the National Academy of Sciences of the United States of America 2011;108(40):16669-74.
18. Andrieu G, Belkina AC, Denis GV. Clinical trials for BET inhibitors run ahead of the science. Drug discovery today Technologies 2016;19:45-50.
19. Donato E, Croci O, Campaner S. Elongation vs stalling: place your BET. Oncotarget 2017;8(67):110737-38.
20. Hogg SJ, Newbold A, Vervoort SJ, Cluse LA, Martin BP, Gregory GP, et al. BET Inhibition Induces Apoptosis in Aggressive B-Cell Lymphoma via Epigenetic
Regulation of BCL-2 Family Members. Molecular cancer therapeutics 2016;15(9):2030-41.
21. Alqahtani A, Choucair K, Ashraf M, Hammouda DM, Alloghbi A, Khan T, et al. Bromodomain and extra-terminal motif inhibitors: a review of preclinical and clinical
advances in cancer therapy. Future science OA 2019;5(3):FSO372.
22. Prochownik EV, Kukowska J, Rodgers C. c-myc antisense transcripts accelerate differentiation and inhibit G1 progression in murine erythroleukemia cells.
Molecular and cellular biology 1988;8(9):3683-95.
23. Sklar MD, Thompson E, Welsh MJ, Liebert M, Harney J, Grossman HB, et al. Depletion of c-myc with specific antisense sequences reverses the transformed phenotype
in ras oncogene-transformed NIH 3T3 cells. Molecular and cellular biology 1991;11(7):3699-710.
24. Kipshidze N, Iversen P, Overlie P, Dunlap T, Titus B, Lee D, et al. First human experience with local delivery of novel antisense AVI-4126 with Infiltrator
catheter in de novo native and restenotic coronary arteries: 6-month clinical and angiographic follow-up from AVAIL study. Cardiovascular revascularization
medicine : including molecular interventions 2007;8(4):230-5.
25. Miller AJ, Chang A, Cunningham PN. Chronic Microangiopathy Due to DCR-MYC, a Myc-Targeted Short Interfering RNA. American journal of kidney diseases : the
official journal of the National Kidney Foundation 2019.
26. Nanbru C, Lafon I, Audigier S, Gensac MC, Vagner S, Huez G, et al. Alternative translation of the proto-oncogene c-myc by an internal ribosome entry site. The
Journal of biological chemistry 1997;272(51):32061-6.
27. Wiegering A, Uthe FW, Jamieson T, Ruoss Y, Huttenrauch M, Kuspert M, et al. Targeting Translation Initiation Bypasses Signaling Crosstalk Mechanisms That
Maintain High MYC Levels in Colorectal Cancer. Cancer discovery 2015;5(7):768-81.
28. Jain S, Wang X, Chang CC, Ibarra-Drendall C, Wang H, Zhang Q, et al. Src Inhibition Blocks c-Myc Translation and Glucose Metabolism to Prevent the Development
of Breast Cancer. Cancer research 2015;75(22):4863-75.
29. Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 2003;22(40):6151-9.
30. Prochownik EV, Vogt PK. Therapeutic Targeting of Myc. Genes & cancer 2010;1(6):650-59.
31. Fletcher S, Prochownik EV. Small-molecule inhibitors of the Myc oncoprotein. Biochimica et biophysica acta 2015;1849(5):525-43.
32. Chauhan J, Wang H, Yap JL, Sabato PE, Hu A, Prochownik EV, et al. Discovery of methyl 4′-methyl-5-(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)-[1,1′-biphenyl]-3-
carboxylate, an improved small-molecule inhibitor of c-Myc-max dimerization. ChemMedChem 2014;9(10):2274-85.
33. Stellas D, Szabolcs M, Koul S, Li Z, Polyzos A, Anagnostopoulos C, et al. Therapeutic effects of an anti-Myc drug on mouse pancreatic cancer. Journal of the
National Cancer Institute 2014;106(12).
34. Hart JR, Garner AL, Yu J, Ito Y, Sun M, Ueno L, et al. Inhibitor of MYC identified in a Krohnke pyridine library. Proceedings of the National Academy of
Sciences of the United States of America 2014;111(34):12556-61.
35. Yu C, Niu X, Jin F, Liu Z, Jin C, Lai L. Structure-based Inhibitor Design for the Intrinsically Disordered Protein c-Myc. Scientific reports 2016;6:22298.
36. Seo HK, Ahn KO, Jung NR, Shin JS, Park WS, Lee KH, et al. Antitumor activity of the c-Myc inhibitor KSI-3716 in gemcitabine-resistant bladder cancer.
Oncotarget 2014;5(2):326-37.
37. Jeong KC, Kim KT, Seo HH, Shin SP, Ahn KO, Ji MJ, et al. Intravesical instillation of c-MYC inhibitor KSI-3716 suppresses orthotopic bladder tumor growth. The
Journal of urology 2014;191(2):510-8.
38. Soodgupta D, Pan D, Cui G, Senpan A, Yang X, Lu L, et al. Small Molecule MYC Inhibitor Conjugated to Integrin-Targeted Nanoparticles Extends Survival in a Mouse
Model of Disseminated Multiple Myeloma. Molecular cancer therapeutics 2015;14(6):1286-94.
39. Jiang H, Bower KE, Beuscher AEt, Zhou B, Bobkov AA, Olson AJ, et al. Stabilizers of the Max homodimer identified in virtual ligand screening inhibit Myc
function. Molecular pharmacology 2009;76(3):491-502.
40. Struntz NB, Chen A, Deutzmann A, Wilson RM, Stefan E, Evans HL, et al. Stabilization of the Max Homodimer with a Small Molecule Attenuates Myc-Driven
Transcription. Cell chemical biology 2019;26(5):711-23 e14.
41. Soucek L, Helmer-Citterich M, Sacco A, Jucker R, Cesareni G, Nasi S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene
1998;17(19):2463-72.
42. Soucek L, Jucker R, Panacchia L, Ricordy R, Tato F, Nasi S. Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer research
2002;62(12):3507-10.
43. Annibali D, Whitfield JR, Favuzzi E, Jauset T, Serrano E, Cuartas I, et al. Myc inhibition is effective against glioma and reveals a role for Myc in proficient
mitosis. Nature communications 2014;5:4632.
44. Sodir NM, Kortlever RM, Barthet VJA, Campos T, Pellegrinet L, Kupczak S, et al. Myc instructs and maintains pancreatic adenocarcinoma phenotype. Cancer
discovery 2020.
45. Soucek L, Nasi S, Evan GI. Omomyc expression in skin prevents Myc-induced papillomatosis. Cell death and differentiation 2004;11(9):1038-45.
46. Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, et al. Modelling Myc inhibition as a cancer therapy. Nature 2008;455(7213):679-83.
47. Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice.
Genes & development 2013;27(5):504-13.
48. Alimova I, Pierce A, Danis E, Donson A, Birks DK, Griesinger A, et al. Inhibition of MYC attenuates tumor cell self-renewal and promotes senescence in SMARCB1-
deficient Group 2 atypical teratoid rhabdoid tumors to suppress tumor growth in vivo. International journal of cancer 2019;144(8):1983-95.
49. Fiorentino FP, Tokgun E, Sole-Sanchez S, Giampaolo S, Tokgun O, Jauset T, et al. Growth suppression by MYC inhibition in small cell lung cancer cells with TP53
and RB1 inactivation. Oncotarget 2016;7(21):31014-28.
50. Beaulieu ME, Jauset T, Masso-Valles D, Martinez-Martin S, Rahl P, Maltais L, et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to
viable anti-MYC therapy. Science translational medicine 2019;11(484).
51. Brown ZZ, Mapelli C, Farasat I, Shoultz AV, Johnson SA, Orvieto F, et al. Multiple Synthetic Routes to the Mini-Protein Omomyc and Coiled-Coil Domain
Truncations. The Journal of organic chemistry 2020;85(3):1466-75.
52. Demma MJ, Hohn MJ, Sun A, Mapelli C, Hall B, Walji A, et al. Inhibition of Myc transcriptional activity by a mini-protein based upon Mxd1. FEBS letters 2020.
53. Huang HL, Weng HY, Wang LQ, Yu CH, Huang QJ, Zhao PP, et al. Triggering Fbw7-mediated proteasomal degradation of c-Myc by oridonin induces cell growth
inhibition and apoptosis. Molecular cancer therapeutics 2012;11(5):1155-65.
54. Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer
cell 2009;15(1):67-78.
55. Macarulla T, Cervantes A, Elez E, Rodriguez-Braun E, Baselga J, Rosello S, et al. Phase I study of the selective Aurora A kinase inhibitor MLN8054 in patients
with advanced solid tumors: safety, pharmacokinetics, and pharmacodynamics. Molecular cancer therapeutics 2010;9(10):2844-52.
56. Beltran H, Oromendia C, Danila DC, Montgomery B, Hoimes C, Szmulewitz RZ, et al. A Phase II Trial of the Aurora Kinase A Inhibitor Alisertib for Patients with
Castration-resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clinical cancer research : an official journal of the American Association for
Cancer Research 2019;25(1):43-51.
57. Falchook G, Coleman RL, Schilder RJ. Paclitaxel and Alisertib in Recurrent Ovarian Cancer-In Reply. JAMA oncology 2019;5(6):910-11.
58. Falchook G, Coleman RL, Roszak A, Behbakht K, Matulonis U, Ray-Coquard I, et al. Alisertib in Combination With Weekly Paclitaxel in Patients With Advanced
Breast Cancer or Recurrent Ovarian Cancer: A Randomized Clinical Trial. JAMA oncology 2019;5(1):e183773.
59. Peter S, Bultinck J, Myant K, Jaenicke LA, Walz S, Muller J, et al. Tumor cell-specific inhibition of MYC function using small molecule inhibitors of the HUWE1
ubiquitin ligase. EMBO molecular medicine 2014;6(12):1525-41.
60. Sears RC. The life cycle of C-myc: from synthesis to degradation. Cell Cycle 2004;3(9):1133-7.
61. Junttila MR, Westermarck J. Mechanisms of MYC stabilization in human malignancies. Cell Cycle 2008;7(5):592-6.
62. Farrell AS, Allen-Petersen B, Daniel CJ, Wang X, Wang Z, Rodriguez S, et al. Targeting inhibitors of the tumor suppressor PP2A for the treatment of pancreatic
cancer. Molecular cancer research : MCR 2014;12(6):924-39.
63. Janghorban M, Farrell AS, Allen-Petersen BL, Pelz C, Daniel CJ, Oddo J, et al. Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proceedings of
the National Academy of Sciences of the United States of America 2014;111(25):9157-62.
64. Sun X, Wang J, Yao X, Zheng W, Mao Y, Lan T, et al. A chemical approach for global protein knockdown from mice to non-human primates. Cell discovery 2019;5:10.
65. Kaini RR, Shen-Gunther J, Cleland JM, Greene WA, Wang HC. Recombinant Xeno-Free Vitronectin Supports Self-Renewal and Pluripotency in Protein-Induced
Pluripotent Stem Cells. Tissue engineering Part C, Methods 2016;22(2):85-90.
66. Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven
tumorigenesis. Science 2012;335(6066):348-53.
67. Hydbring P, Larsson LG. Cdk2: a key regulator of the senescence control function of Myc. Aging 2010;2(4):244-50.
68. Huang CH, Lujambio A, Zuber J, Tschaharganeh DF, Doran MG, Evans MJ, et al. CDK9-mediated transcription elongation is required for MYC addiction in
hepatocellular carcinoma. Genes & development 2014;28(16):1800-14.
69. Horiuchi D, Camarda R, Zhou AY, Yau C, Momcilovic O, Balakrishnan S, et al. PIM1 kinase inhibition as a targeted therapy against triple-negative breast tumors
with elevated MYC expression. Nature medicine 2016;22(11):1321-29.
☞ 자세한 내용은 내용바로가기 또는 첨부파일을 이용하시기 바랍니다.