

기술동향
딥러닝: 유전체학을 위한 새로운 컴퓨터 모델링 기술
- 등록일2021-01-26
- 조회수6866
- 분류기술동향
-
자료발간일
2021-01-21
-
출처
생물학연구정보센터(BRIC)
- 원문링크
-
키워드
#딥러닝#유전체학#염기서열
- 첨부파일
 pdf_0003685.pdf
pdf_0003685.pdf
딥러닝: 유전체학을 위한 새로운 컴퓨터 모델링 기술
요약문
데이터 주도 과학으로서 유전체학 분야에서는 기계학습을 데이터 특성 파악 및 새로운 생물학적 가설 유도를 위하여 이용되고 있다. 하지만, 급격하게 증가하고 있는 유전체 데이터에서 새로운 의미들을 얻기 위해서는 보다 심층적인 기계학습 모델을 필요로 하는데, 컴퓨터 비젼과 자연어 처리 분야에서 주로 사용되는 딥러닝 기술이 이제는 생물학 분야 중에서도 DNA accessibility와 Splicing과 같은 유전자 조절 기전에 있어서 유전적 변이의 영향을 예측하는 등의 수많은 유전체학 데이터 모델링을 위한 방법으로서 사용되고 있다.
목 차
1. 서론
2. 지도 학습(Supervised learning)
2.1. DNN을 이용한 복잡도 의존성 모델링
2.2. 컨볼루션을 통한 Sequential 데이터 내 국소 패턴 발굴(CNN in genomics)
2.3. 순환신경망을 이용한 장거리 염기서열 데이터 모델링(RNN in genomics)
2.4. 그래프-CNN을 이용한 그래프 구조 데이터 모델링
2.5. 연산 수행 사이의 정보 공유 및 다중 데이터 통합
2.6. 전이학습을 통합 소규모 데이터 기반 모델 훈련
3. 예측 설명
3.1. 특성 중요도 파악을 통한 입출력 관계 파악
3.2. 염기서열 Motif 발굴
3.3. 해석 가능한 파라미터와 활성함수를 포함한 인공신경망
4. 비지도적 학습
5. 유전체학에서의 딥러닝의 영향
6. 결론 및 전망
1. 서론
기능적 혹은 광범위한 개념의 유전체학은 유기체의 유전체적 요소들의 기능을 규명하는데, genome 시퀀싱, 전사체 분석, 단백체학과 같은 유전체 스케일에서의 실험을 통하여 이루어진다. 유전체학은 데이터 주도하는 과학으로서 발전하고 있는데, 이는 기존의 지식을 기반으로 하는 모델이나 가설에 대한 연구보다는 새로운 정보들을 발굴하는 것을 시사한다. 대표적인 예로는 유전자형과 표현형 사이의 관련성, 환자들의 분류를 위한 바이오마커 발굴, 전사 강화인자(enhancer)와 같은 생화학 활성이 이루어진 genome 상의 위치, 유전자들의 기능성 예측 등이 있다.
유전체 데이터는 상당히 규모가 크고 구조가 복잡하기 때문에, 새로운 발견과 밝혀지지 않은 상관성 및 연계성을 찾고 새로운 가설 및 모델을 제시하여 예측하기 위해서는 전문적인 분석 기술 도구가 필요하다. 여러 분석 알고리즘들 중, 가정과 특정 도메인들을 사용자가 직접 설정해야 하는 기법들과는 달리, 기계학습에서는 데이터 내에서의 특징 및 패턴을 자동적으로 발견하기에, 유전체학과 같이 데이터가 주도하는 분야에서 사용하기에 적합하다. 하지만, 기계학습 방법의 단점은 보여지는 데이터 자체에 매우 의존도가 높으며, 특징 추출 과정에서 어떻게 특징들을 추출하는지, 어떤 특징들이 기계학습을 이용한 예측에 사용되는지에 따라 성능이 감소될 수 있다. 위와 같은 한계점을 딥러닝에서는 DNN (Deep Neural Network)의 발전을 통하여 해결하였다. DNN은 수행과정 중에 얻게 되는 중간결과들을 입력값으로 다시 사용하여 이를 컴퓨터가 스스로 반복하여 계산하는 방식인 end-to-end 학습으로 복잡한 특징들을 계산하고 추출하는데 성공적인 결과를 보여주었다.
DNN의 구축과 훈련은 데이터의 폭발적인 증가세와 알고리즘의 발전 그리고 GPU와 같은 컴퓨터 하드웨어 요소의 발전이 기반이 되어 가능하였다. 지난 7년간, DNN은 컴퓨터 비전, 음성 인식 등 컴퓨터 과학 분야에서 주로 사용되었는데, 2015년 DNA 염기서열 데이터에 처음으로 적용된 연구가 발표되면서부터, DNN 및 딥러닝 기술을 유전체학 분야에 적용한 수많은 논문들이 게재되었으며, 동시에 딥러닝 분야의 연구자들은 위의 기술들의 성능 향상과 모델링 기술 레퍼토리의 확대에 힘쓰고 있어, 이들 중 일부 기술들은 이미 유전체 연구분야에서 영향력을 보이고 있다.
본 리뷰에서 우리는 딥러닝을 이용한 모델링 기술과 유전체 데이터로의 적용에 대하여 우선하여, 지도적 학습을 위한 4가지의 주요 모델들과 함께 이 방법들이 어떻게 유전체 데이터 내에서 패턴을 찾아내는지를 설명할 것이다. 그 다음으로 여러 가지 데이터셋과 데이터 유형들의 통합을 위한 multitask/ multimodal 학습 기술과 기존에 존재하는 모델을 기반으로 빠르게 변형 및 발전 시켜 사용하는 전이 학습 기술, 마지막으로 비지도적 학습을 위하여 사용되는 autoencoder와 단세포 유전체학 분야에서 처음으로 적용된 generative adversarial networks (GANs) 알고리즘에 대하여 소개할 것이다. 유전체학 분야를 넘어서 딥러닝에 대한 보다 깊고 자세한 배경지식들을 위해서는 이 논문과 더불어 컴퓨터 생물학자 및 생물정보학자들을 대상으로 하는 딥러닝 관련 다른 여러 논문들을 함께 읽어보는 것을 권장한다.
2. 지도 학습(Supervised learning)
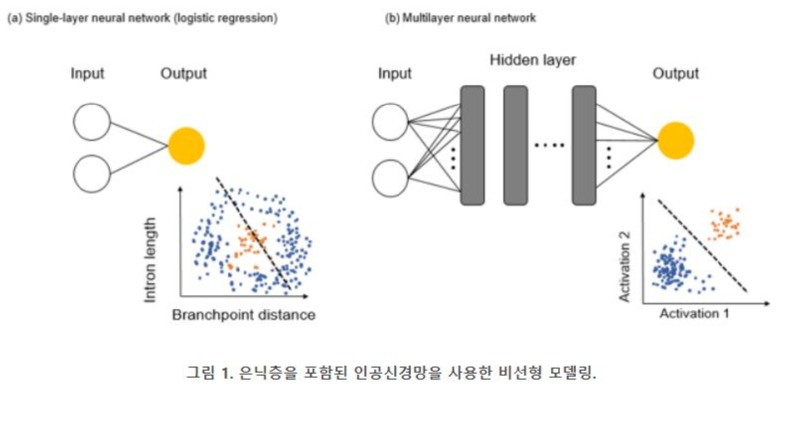
지도학습의 목적은 특징들을 입력값으로 받아 타겟 변수에 대한 예측치를 결과로 반환하는 모델을 얻는 것이다. 예를 들어, splicing 현상에서, RNA 염기서열들을 특징으로서 사용하고 이를 입력 값으로 하여 특정 intron이 spliced out인지 아닌지를 설계한 모델을 훈련하여 예측하는 것이 지도학습을 통하여 해결할 수 있는 문제 중 하나이다 (그림 1). 여기서 기계학습 모델을 훈련시킨다는 것은 모델을 구성하고 있는 여러 가지 파라미터(옵션값)들을 학습하면서 손실함수(또는 비용함수)를 최소화 시키는 과정을 의미한다
2.1. DNN을 이용한 복잡도 의존성 모델링
컴퓨터 생물학 분야에서의 많은 지도적 학습 문제들의 경우, 입력 데이터를 행렬 형식으로 받는데, 데이터가 생성될 때부터 행렬 형태로 주어지는 경우도 있지만, 전처리 과정을 통하여 행렬화시켜야 하는 경우도 있다(DNA 염기서열의 K-mer 개수로의 형태 변환). Intron-Splicing 예측 문제에서는, intron 길이와 splicing branchpoint 위치가 전처리된 특징들로써 행렬 데이터에 수집된다. 이와 같이 행렬(표) 형식의 데이터는 단순한 선형 모델(예: 로지스틱 회귀)부터 보다 일반적이고 유연한 모델인 비선형 모델들(예: 신경망, 비선형 회귀)에서 표준적으로 사용되는 데이터 포맷이다. 위와 같은 입력 데이터 형태(표 형식)로 Intron-Splicing 문제에 대하여 단순 선형 회귀 분석법인 로지스틱 회귀 분석(이진 분류기)과 DNN (비선형 지도학습법)을 적용하여 예측 결과를 통한 성능을 비교해본 결과, 단순히 입력 값에 있는 특징들의 가중치를 기반으로 한 가중치 합만으로는 intron spliced out 여부에 대한 예측을 정확히 할 수 없는데 반하여, DNN (다중층 신경망)을 이용한 경우 명료하게 분류해낼 수 있음 보여준다 (그림 1).
그림 1B에서 보인 신경망을 이용한 예측분석은 여러 은닉층(hidden layer)들을 사용하여 자동적인 비선형적 특성들을 학습하여 단순 선형회귀법(로지스틱 회귀)보다 좋은 성능을 보였는데, 이때 설계된 여러 은닉층들을 다수의 선형 모델들이라고 생각할 수 있으며, 이들의 결과값들이 비선형 활성함수(예, Sigmoid, ReLu 함수)에 의하여 변형된다. 즉, 이러한 은닉층들은 입력 특성들을 통해 데이터의 복잡한 패턴들을 파악해 분류 문제를 해결하는데 활용된다.
DNN에서는 많은 은닉층들을 활용하고 은닉층의 각 뉴런이 이전 은닉층의 모든 뉴런으로부터 입력을 받는 구조를 “fully connected”라고 한다. 신경망은 일반적으로 “stochastic gradient descent“라는 알고리즘을 모델 트레이닝에 사용하는데, 이는 데이터셋의 규모가 충분히 큰 경우에 많이 파라미터 학습법이다. Fully connected 신경망(Fully connected neural network; FCNN)은 이미 유전체학 분야에서 많이 사용되어지고 있는데, splicing 현상에서 특정 exon이 spliced-in 될 확률, 주어진 질병 유발 유전적 변이의 우선순위 예측, 주어진 genomics region 내에서의 cis 조절인자 예측이 대표적인 적용사례이다.
물론 FCNN 방법이 항상 좋은 성능을 보이는 것은 아니다, 전통적인 기계학습 알고리즘인 random forest 혹은 선형회귀보다 성능이 좋다는 보고가 많이 따르지만, 어떠한 경우는 gradient-boosted decision tree가 FCNN의 성능을 앞선다는 결과가 Kaggle 대회에서 보인 바 있다. 그럼에도 FCNN은 딥러닝 툴박스로서 매우 중요한 요소이며, 다른 신경망 층(예: convolutional layer)과도 효율적으로 병합되어 사용되기도 한다.
2.2. 컨볼루션을 통한 Sequential 데이터 내 국소 패턴 발굴(CNN in genomics)
데이터 유형 특이적 패턴 파악(데이터 특이적 국소 패턴 분석)은 효율적인 예측을 위해서 중요한데, 단일 유형이 아닌 다양한 형태의 데이터가 존재하는데 이들의 데이터가 무작위로 섞인 상태로 행렬화되어 FCNN을 적용한다면 데이터 내의 유의미한 패턴을 파악하기 어려울 것이다. 유전체학 분야의 한 가지 예를 들자면, 특정 genomic region이 주어질 때, 해당 영역이 전사인자가 붙는 구간인지 아닌지를 예측하는 문제를 들 수 있다. 데이터는 ChIP-seq을 통해 얻어진 높은 신뢰도를 가진 특정 genomic region에 대한 binding event 데이터 한 가지와 전사인자가 붙는 구간의 시퀀스(binding motif)이다. Binding event는 숫자형 데이터이고, Binding motif는 시퀀스 데이터이다. K-mer instance의 개수를 세거나, 해당 시퀀스 내에서 position weight matrix (PWM)을 사용한다면 가능할 수도 있겠지만, 전사인자가 여러 개의 motif 조합에 따른 영향을 받는다는 문제와 과적합(overfitting)이라는 한계점이 있다.
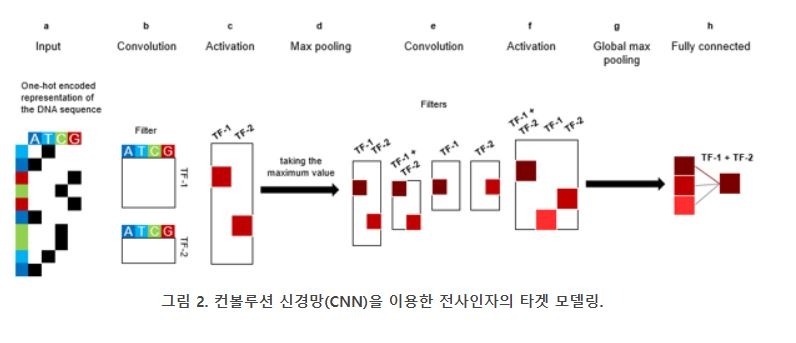
위의 문제의 해결법 중 한 가지가 컨볼루션을 이용하는 것이다. 그림 2에서처럼 컨볼루션 층은 데이터의 국소영역에 적용되어 패턴을 분석한다. 이러한 접근법은 다수의 PWM을 사용하여 염기서열을 스캐닝하는 것으로 보여지기도 한다. 그림 2B에서처럼 컨볼루션 층에서는 전체 염기서열에 걸쳐 여러 개의 필터를 이용하여 스캐닝을 수행하고, 비선형 활성 함수(예: ReLU)를 필터링한 결과값에 적용시킨다. 활성함수를 통과한 결과값들은 다음으로 “풀링(Pooling)” 연산을 거치게 된다.
풀링 연산은 보통 최대값 혹은 평균값을 이용하여 진행하는데 이 과정을 통하여 데이터 사이즈를 효과적으로 감소시킬 수 있다 (그림 2D). 위의 과정을 통하여, 특정 거리범위 내에서 어떠한 전사인자의 binding motif가 존재하는지를 예측할 수 있다. 이와 같이, 컨볼루션 층의 결과값은 FCNN의 입력 값으로 다시 반환되어 사용되어 최종 예측 과정을 수행해낼 수 있다 (그림 2 F-H). 따라서, 한 개의 딥러닝 모델 안에서 서로 다른 유형의 신경망층(예, 컨볼루션 층 + Fully connected 층)이 혼재되어 좋은 성능을 보일 수 있는데, 유전체학 분야에서는 특히 염색체 구조 관련 분석과 같이 공간적 특성이 반영된 염기서열 데이터 분석 관련하여 적용할 수 있다.
...................(계속)
☞ 자세한 내용은 내용바로가기 또는 첨부파일을 이용하시기 바랍니다.
관련정보
지식
동향